42 Adding a Latent Stage to Make an SEIR Model
EpiModel includes an integrated SIR model, but here we show how to model an SEIR disease, one with a latent (exposed) stage between infection and infectiousness, as many respiratory infections have. The E compartment is an exposed state in which a person has been infected but is not infectious to others. Some infectious diseases have this latent non-infectious stage, and in general it provides a general framework for transmission risk that is dependent on one’s stage of disease progression. The parameters used below are stylized to demonstrate the extension API clearly rather than calibrated to any specific pathogen, and the time step is an abstract unit; for a real analysis you would substitute published estimates and fix the time step (for example, a day or a week).
This tutorial has two R scripts you should download: a primary script containing the code below and a separate module script. Download both and put them in the same working directory.
We build this SEIR module by hand because writing one module yourself is the fastest way to internalize the extension API. The same model, parameterized to provide both SEIR and SEIRS from a single module, is maintained as a Gallery example: SEIR/SEIRS: Adding an Exposed State. That is the copy you would actually reuse and extend, and the lab works directly with it. The get_/set_ accessors and control.net settings used below are documented in full in the EpiModel package vignettes at epimodel.org.
42.1 Setup
First start by loading EpiModel and clearing your global environment.
42.2 EpiModel Model Extensions
42.2.1 Conceptualization
The first step to any EpiModel extension model is to conceptually identify what new functionality, above and beyond the built-in models, is desired and where that functionality should be added. There are often many “right” answers to these questions, and this aspect is only learned over time. But in general, it is helpful to map out a model extension on a state/flow diagram to pinpoint where the additions should be concentrated.
42.2.1.1 SIR Module Set
In this particular model, we will be adding a new disease state that occurs in between the susceptible disease state and an infectious disease state in an SIR model. The standard, built-in SIR model in EpiModel uses a set of modules (elements of the model), each with its own associated function (the realization of those elements in code). To ground the discussion, let’s fit and run a basic built-in SIR model and print the result.
Code
nw <- network_initialize(n = 100)
formation <- ~edges
target.stats <- 50
coef.diss <- dissolution_coefs(dissolution = ~offset(edges), duration = 20)
est1 <- netest(nw, formation, target.stats, coef.diss, verbose = FALSE)
param <- param.net(inf.prob = 0.3, rec.rate = 0.1)
init <- init.net(i.num = 10, r.num = 0)
control <- control.net(type = "SIR", nsteps = 25, nsims = 1, verbose = FALSE)
mod1 <- netsim(est1, param, init, control)
mod1EpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type: SIR
No. simulations: 1
No. time steps: 25
No. NW groups: 1
Fixed Parameters
---------------------------
inf.prob = 0.3
rec.rate = 0.1
act.rate = 1
groups = 1
Model Output
-----------------------
Variables: s.num i.num r.num num si.flow ir.flow
Networks: sim1
Transmissions: sim1
Formation Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 50 48.32 -3.36 3.822 -0.44 NA 6.115
Duration Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 20 18.105 -9.476 0.329 -5.757 NA 1.113
Dissolution Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.05 0.05 0.149 0.007 0.011 NA 0.034Printing a built-in model reports its type, run settings, and the compartments and flows it tracked, but not the list of module functions: for a built-in model EpiModel selects those internally from type. Two things do expose the module set. The control.net argument list (below) names every module slot, and once we build our own extension model later in this tutorial (with type = NULL), printing it adds a Model Functions section that lists exactly the modules that ran.
42.2.1.2 Module Classes
This includes a series of modules, which we classify into Standard and Flexible modules as shown on the table below. Any modules may be modified, but standard modules are those that are typically not modified because they generalize the core internal processes for simulation object generation, network resimulation, and epidemic bookkeeping. Flexible modules, in contrast, are those that are likely necessary to modify for an EpiModel extension.
In the example above, we used all of these modules except the arrivals and departures modules because we had a closed population. In addition to these set of built-in modules, any user can add more modules to this set, depending on what is needed. Again, this involves some conceptualization of how to organize the model processes, including whether those processes are similar to the built-in modules or something new.
42.2.1.3 Modules vs. Functions
All modules have associated functions, and these are passed into the epidemic model run in netsim through control.net. Printing out the arguments for this function, you will see that each of the standard modules have default associated functions as inputs, and flexible modules have a default of NULL.
function (type, nsteps, start = 1, nsims = 1, ncores = 1, resimulate.network = FALSE,
tergmLite = FALSE, cumulative.edgelist = FALSE, truncate.el.cuml = 0,
attr.rules, epi.by, initialize.FUN = initialize.net, resim_nets.FUN = resim_nets,
summary_nets.FUN = summary_nets, infection.FUN = NULL, recovery.FUN = NULL,
departures.FUN = NULL, arrivals.FUN = NULL, nwupdate.FUN = nwupdate.net,
prevalence.FUN = prevalence.net, verbose.FUN = verbose.net,
module.order = NULL, save.nwstats = TRUE, nwstats.formula = "formation",
save.transmat = TRUE, save.network, save.run = FALSE, save.cumulative.edgelist = FALSE,
save.other, verbose = TRUE, verbose.int = 1, skip.check = FALSE,
raw.output = FALSE, future.use.plan = FALSE, tergmLite.track.duration = FALSE,
set.control.ergm = control.simulate.formula(MCMC.burnin = 2e+05),
set.control.tergm = control.simulate.formula.tergm(MCMC.maxchanges = .Machine$integer.max),
save.diss.stats = TRUE, dat.updates = NULL, ...)
NULLFor built-in models, EpiModel selects which modules are needed based on the model parameters and initial conditions. For each time step, the modules run in the order in which they are specified in the output here, which for a built-in model matches the order in which they are listed in control.net. Extension models add one wrinkle we will meet below: a module passed under a name that is not one of control.net’s own arguments is a user module, and EpiModel runs all user modules before all built-in ones, whatever order they were written in.
42.2.1.4 Built-in Functions as Templates
Any of the built-in functions associated with flexible modules are intended to be templates for user inspection and extension for research-level models. A built-in SIR model uses infection.net as its infection module. That function has a help page briefly describing what it does. And you can also inspect the function contents with:
We’ll use an edited down version of that function, with some additional explanation, below. In addition, the disease progression state transition within an SIR model (and an SIS model too) is handled by the recovery module, with an associated recovery.net function.
We will use an edited down version of that function as a template for a new, more general disease progression module.
42.2.2 The EpiModel API
All EpiModel module functions have a set of shared design requirements. This set of requirements defines the EpiModel Application Programming Interface (API) for extension. The best way to learn this is through a concrete example like this, but here are the general API rules:
- Each module has an associated R function, the general design of which is:
The function takes an object called dat, which is the master data object passed around by netsim, performs some processes (e.g., infection, recovery, aging, interventions), updates the dat object, and then returns that object. The other input argument to each function must be at, which is a time step counter.
Data are stored on the
datobject in a particular way: in sublists that are organized by category. The main categories of data to interact with include model inputs (parameters, initial conditions, and controls) from those three associated input functions; nodal attributes (e.g., an individual disease status for each person); and summary statistics (e.g., the disease prevalence at at time step). There are accessor functions for reading (these are theget_functions) and writing (these are theset_functions) to thedatobject in the appropriate place.The typical function design involves three steps: a) reading the relevant inputs from the
datobject; b) performing some micro-level process on the nodes that is usually a function of fixed parameters and time-varying nodal attributes; c) writing the updated objects back on to thedatobject.
Let’s see how this API works by extending our infection and recovery functions to transition an SIR model into an SEIR model.
42.2.3 Infection Module
The built-in infection module for an SIR model performs the following functions listed in the function below. The core process is determining which edges are eligible for a disease transmission to occur, and then randomly simulating that transmission process. Why is it necessary to update the infection function for an SEIR model? Because an SIR involves a transition between S and I disease statuses when a new infection occurs, but an SEIR model involves a transition between S and E disease statuses. It’s a small, but important change. Here’s the full modified function, with embedded comments. Note that we can use the browser function to run this function in debug mode by uncommenting the third line (we will demonstrate this).
Code
infect <- function(dat, at) {
## Uncomment this to run environment interactively
# browser()
## Attributes ##
active <- get_attr(dat, "active")
status <- get_attr(dat, "status")
infTime <- get_attr(dat, "infTime")
## Parameters ##
inf.prob <- get_param(dat, "inf.prob")
act.rate <- get_param(dat, "act.rate")
## Find infected nodes ##
idsInf <- which(active == 1 & status == "i")
nActive <- sum(active == 1)
nElig <- length(idsInf)
## Initialize default incidence at 0 ##
nInf <- 0
## If any infected nodes, proceed with transmission ##
if (nElig > 0 && nElig < nActive) {
## Look up discordant edgelist ##
del <- discord_edgelist(dat, at)
## If any discordant pairs, proceed ##
if (!(is.null(del))) {
# Set parameters on discordant edgelist data frame
del$transProb <- inf.prob
del$actRate <- act.rate
del$finalProb <- 1 - (1 - del$transProb)^del$actRate
# Stochastic transmission process
transmit <- rbinom(nrow(del), 1, del$finalProb)
# Keep rows where transmission occurred
del <- del[which(transmit == 1), ]
# Look up new ids if any transmissions occurred
idsNewInf <- unique(del$sus)
nInf <- length(idsNewInf)
# Set new attributes and transmission matrix
if (nInf > 0) {
status[idsNewInf] <- "e"
infTime[idsNewInf] <- at
dat <- set_attr(dat, "status", status)
dat <- set_attr(dat, "infTime", infTime)
dat <- set_transmat(dat, del, at)
}
}
}
## Save summary statistic for S->E flow
dat <- set_epi(dat, "se.flow", at, nInf)
return(dat)
}Each step is relatively self-explanatory with the comments, but we will run this interactively at the end of this tutorial to step through the updated data structures. Infection is one of the more complex processes because it involves a dyadic process (a contact between an S and I node). That involves a construction of a discordant edgelist; that is, a list of edges in which there is a disease discordant dyad.
The main point here is: we have made a change to the infection module function, and it consists of updating the disease status of a newly infected persons to "e" instead of "i". Additionally, we are tracking a new summary statistic, se.flow that tracks the size of the flow from S to E based on the number of new infected, nInf at the time step.
42.2.4 Progression Module
Next up is the disease progression module. Here, we have generalized the built-in recovery module function to handle two disease progression transitions after infection: E to I (latent to infectious stages) and I to R (infectious to recovered stages). Like many individual-level transitions, this involves flipping a weighted coin with rbinom: this performs a series of random Bernoulli draws based on the specified parameters. Here is the full model function.
Code
progress <- function(dat, at) {
## Uncomment this to function environment interactively
# browser()
## Attributes ##
active <- get_attr(dat, "active")
status <- get_attr(dat, "status")
## Parameters ##
ei.rate <- get_param(dat, "ei.rate")
ir.rate <- get_param(dat, "ir.rate")
## E to I progression process ##
nInf <- 0
idsEligInf <- which(active == 1 & status == "e")
nEligInf <- length(idsEligInf)
if (nEligInf > 0) {
vecInf <- which(rbinom(nEligInf, 1, ei.rate) == 1)
if (length(vecInf) > 0) {
idsInf <- idsEligInf[vecInf]
nInf <- length(idsInf)
status[idsInf] <- "i"
}
}
## I to R progression process ##
nRec <- 0
idsEligRec <- which(active == 1 & status == "i")
nEligRec <- length(idsEligRec)
if (nEligRec > 0) {
vecRec <- which(rbinom(nEligRec, 1, ir.rate) == 1)
if (length(vecRec) > 0) {
idsRec <- idsEligRec[vecRec]
nRec <- length(idsRec)
status[idsRec] <- "r"
}
}
## Write out updated status attribute ##
dat <- set_attr(dat, "status", status)
## Save summary statistics ##
dat <- set_epi(dat, "ei.flow", at, nInf)
dat <- set_epi(dat, "ir.flow", at, nRec)
dat <- set_epi(dat, "e.num", at,
sum(active == 1 & status == "e"))
dat <- set_epi(dat, "r.num", at,
sum(active == 1 & status == "r"))
return(dat)
}This set of two progression processes involves querying who is eligible to transition, randomly transitioning some of those eligible, updating the status attribute for those who have progressed, and then recording some new summary statistics.
The two progression steps run in sequence on the same local status vector. The E-to-I block updates status in place, and then the I-to-R block queries which(status == "i"), which now includes the nodes that just became infectious. A node can therefore move E to I and then I to R within a single step, spending no full step as infectious. With small rates this is rare (roughly ei.rate * ir.rate per eligible node), and treating each step’s eligibility as “whoever is in the compartment right now” is a common, defensible choice in discrete-time models. If instead you want each node to make at most one transition per step, snapshot status at the top of the function and query that snapshot in both blocks. Neither version is wrong; the point is that update order within a module is part of the model you are specifying, just as the order between modules is.
42.3 Network Model
With the epidemic modules defined, we will now step back to parameterize, estimate, and diagnose the TERGM. Here we will use a relatively basic model with an edges and degree(1). Here we are not using any nodal attributes in either the TERGM or the epidemic modules, but these could be added (we will get more practice with that tomorrow). Note that we are using a relatively high mean degree (2 per capita) compared to some of our prior models, with lower than expected isolates.
Code
# Initialize the network
nw <- network_initialize(500)
# Define the formation model: edges + degree terms
formation = ~edges + degree(1)
# Input the appropriate target statistics for each term
target.stats <- c(500, 180)
# Parameterize the dissolution model
coef.diss <- dissolution_coefs(dissolution = ~offset(edges), duration = 25)
coef.dissDissolution Coefficients
=======================
Dissolution Model: ~offset(edges)
Target Statistics: 25
Crude Coefficient: 3.178054
Mortality/Exit Rate: 0
Adjusted Coefficient: 3.178054Next we fit the network model.
And then diagnose it. We are including a wide range of degree terms to monitor so we can see the full degree distribution . Although the degree(1) term does not look great visually, it is only off by a few nodes in absolute terms (degree(1) counts nodes with exactly one partner, not edges).
Code
Network Diagnostics
-----------------------
- Simulating 10 networks
- Calculating formation statisticsEpiModel Network Diagnostics
=======================
Diagnostic Method: Dynamic
Simulations: 10
Time Steps per Sim: 500
Formation Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 500 500.491 0.098 1.852 0.265 5.547 21.351
degree0 NA 50.984 NA 0.381 NA 0.966 7.410
degree1 180 174.934 -2.815 0.589 -8.601 2.881 11.313
degree2 NA 115.042 NA 0.404 NA 2.085 9.501
degree3 NA 83.826 NA 0.360 NA 1.823 8.564
degree4 NA 45.431 NA 0.289 NA 1.347 6.756
degree5 NA 19.851 NA 0.205 NA 0.687 4.755
degree6 NA 7.008 NA 0.111 NA 0.297 2.753
degree7 NA 2.159 NA 0.063 NA 0.148 1.556
Duration Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 25 25.048 0.191 0.098 0.49 0.399 1.088
Dissolution Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.04 0.04 -0.023 0 -0.073 0 0.009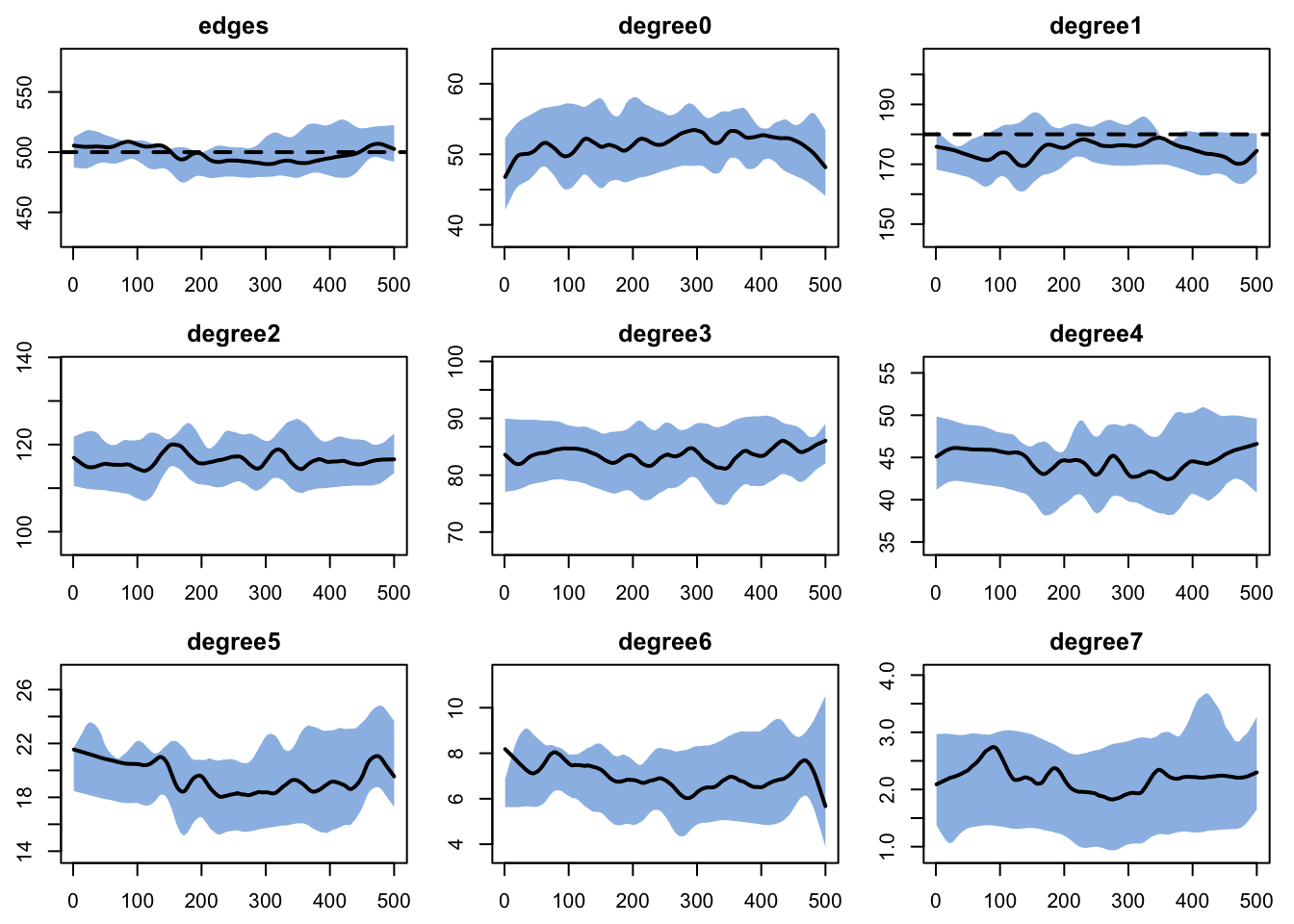
42.4 Epidemic Model Parameterization
The epidemic model parameterization for any extension model consists of using the same three input functions as with built-in models.
First we start with the model parameters in param.net. Note that we have two new rates here: ei.rate and ir.rate. These control the transitions from E to I, and then I to R.
- 1
-
The two standard parameters, read inside
infectbyget_param(dat, "inf.prob")andget_param(dat, "act.rate"). - 2
-
The two new rates. These names are ones we chose; EpiModel does not know them and does not supply them. The same strings appear in the
get_param()calls insideprogress, and that correspondence is the only thing linking the parameter you set here to the value the module reads. A mismatch is not silent:get_param()stops with “There is no parameter called …”, so if you see that error, align the string here with the string in the module function.
Next we specify the initial conditions. Here we are specifying that there are 10 infectious individuals at the epidemic outset. If we wanted to initialize the model with persons only in the latent stage, we would need to set disease status as a nodal attribute on the network instead (following the same approach in the Serosorting Tutorial in Module 7).
Finally, we specify the control settings in control.net. This is where an extension model differs most from a built-in one, so it is worth taking the arguments one at a time. The control.net Guide covers the full argument list.
Code
- 1
-
Source the module functions first.
control.netstores the function objects, captured as they exist at this moment, soinfectandprogresshave to be defined before this call. - 2
-
typeis set toNULLin any extension model, because we are no longer asking EpiModel to pre-select which modules to run; the assembly is manual. This is not merely conventional: EpiModel stops with an error iftypeis non-NULLwhile any user module is present, andprogress.FUNbelow is a user module. - 3
-
infection.FUNis the slot for the infection process, and it defaults toNULL. On the built-in path EpiModel fills it fromtype; because we settype = NULL, nothing fills it, so supplying our owninfectis required rather than optional. Without it the model would run no transmission at all. - 4
-
progress.FUNadds a new module.control.nethas no argument by that name, so it arrives through...as a user module, and EpiModel runs every user module before every built-in module.progresstherefore fires beforeinfectat each time step, whatever order we write them in here. One visible consequence: a node exposed at step t cannot progress out of E until step t+1, becauseprogresshas already run by the timeinfectcreates it. Replacing the built-in recovery module instead would not be equivalent, sincerecovery.FUNis a built-in slot and would run after infection. Use themodule.orderargument if you need to control this directly. - 5
-
resimulate.networkis set toFALSEexplicitly. This is the default, so the line is not necessary, but it is a reminder that this is a model without network feedback.
In the R scripts that we include with this tutorial, you will see that we have two separate R script files (this rendered page shows the function definitions inline for reading, but it still sources them from the separate mod9-SEIR-fx.R, the same split as the downloadable scripts). One file contains the module functions, and the other contains all the other code to parameterize and run the model. We do this because it allows for easier interaction with the functions in browser mode. I will demonstrate this live next. But in general, placing the functions in a separate file conceptually disentangles the model functionality from the model parameterization. One practical consequence of the sourcing rule above: if you edit a module function, re-sourcing the file is not enough on its own, because control.net already captured the old version. Rebuild the control object as well.
42.5 Epidemic Simulation
Finally we are ready to do the epidemic model simulation and analysis. This is done using the same approach as the built-in models. We will start with running one model in the browser mode interactively.
And then go back, comment out the browser lines, re-source the functions, and run a full-scale model with 10 simulations.
Once the model simulation is complete, we can work with the model object just like a built-in model. Start by printing the output to see what is available. Because this is an extension (type = NULL), the printout also includes the Model Functions section promised earlier, listing exactly the modules that ran, our infect and progress alongside the standard bookkeeping modules.
EpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type:
No. simulations: 10
No. time steps: 500
No. NW groups: 1
Fixed Parameters
---------------------------
inf.prob = 0.5
act.rate = 2
ei.rate = 0.01
ir.rate = 0.01
groups = 1
Model Functions
-----------------------
initialize.FUN
resim_nets.FUN
summary_nets.FUN
infection.FUN
nwupdate.FUN
prevalence.FUN
verbose.FUN
progress.FUN
Model Output
-----------------------
Variables: s.num i.num num ei.flow ir.flow e.num r.num
se.flow
Networks: sim1 ... sim10
Transmissions: sim1 ... sim10
Formation Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 500 501.438 0.288 1.772 0.811 8.133 21.394
degree1 180 174.926 -2.819 0.644 -7.885 3.639 11.601
Duration Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 25 24.987 -0.052 0.091 -0.142 0.308 1.059
Dissolution Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.04 0.04 0.179 0 0.58 0 0.009Here is the default plot with all the compartment sizes over time. This includes the new summary statistics we tracked in the disease progression function.
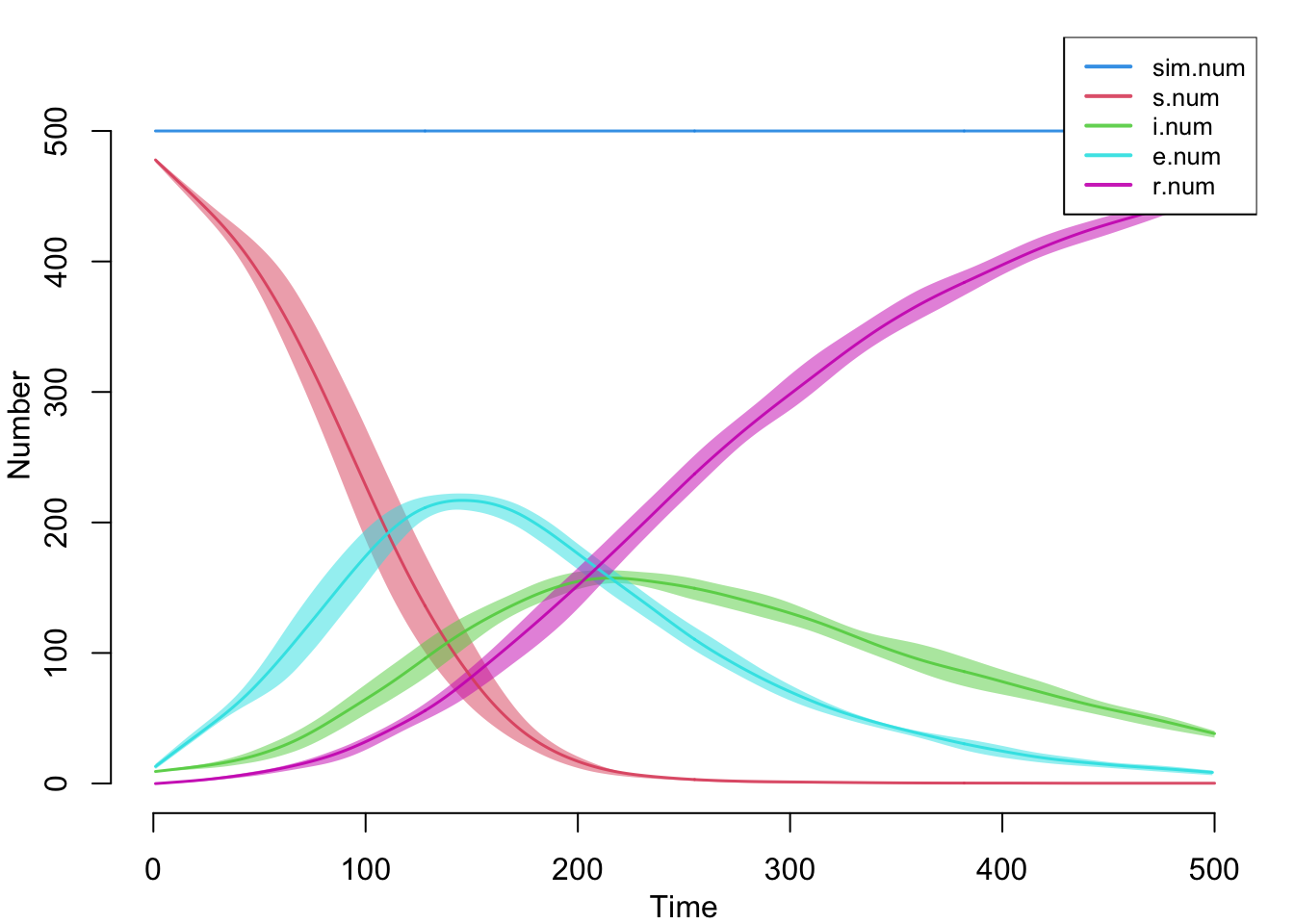
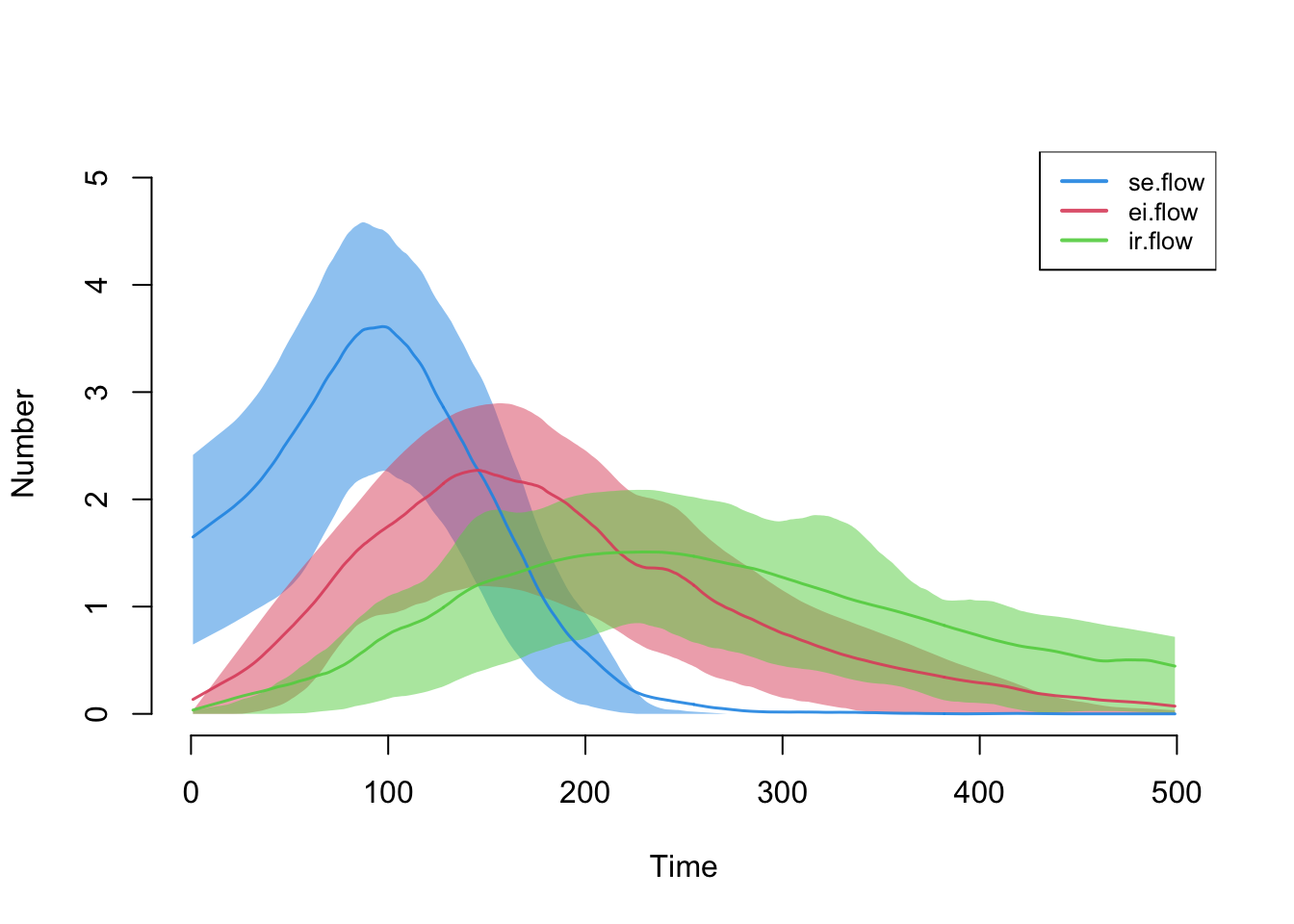
Here are the flow sizes, including the new se.flow incidence tracker that we established in the new infection function.
Finally, here is the data frame output from the model, with rows limited to time step 100 across all 10 simulations.
sim time s.num i.num num ei.flow ir.flow e.num r.num se.flow
100 1 100 182 76 500 5 0 206 33 3
600 2 100 186 76 500 1 1 200 36 2
1100 3 100 231 67 500 3 1 170 27 5
1600 4 100 185 81 500 4 0 197 31 6
2100 5 100 218 60 500 2 1 185 35 2
2600 6 100 214 76 500 2 0 176 29 5
3100 7 100 280 52 500 0 0 145 23 0
3600 8 100 202 88 500 2 0 182 24 4
4100 9 100 150 91 500 0 1 216 39 4
4600 10 100 233 61 500 3 0 170 28 8The transmission matrix records, for each newly infected node, an attributed source (its infector). Below we plot a few of the transmission trees; each traces the onward infections from one initially infected node. A large epidemic descends from a handful of seeds into a few very large trees, so we show a few small-to-moderate ones rather than all of them, to keep the branching legible.
Code
tm1 <- get_transmat(sim)
# get_transmat() returns at most one infector per newly infected node per step:
# if a susceptible had several infected partners transmit in the same step,
# EpiModel resolves that to a single attributed source (chosen at random by
# default, via the deduplicate argument). So "who infected whom" here is one
# plausible source per infection, not a claim that we observed the true one. In
# this SEIR model each node is infected only once, so tm1 is already a clean
# forest; we then plot only the three smallest trees that have at least three
# nodes, skipping the trivial one- and two-node ones, so the branching stays
# legible.
tmf <- tm1
tree_size <- function(root) {
ids <- root
repeat {
kids <- tmf$sus[tmf$inf %in% ids]
if (all(kids %in% ids)) break
ids <- union(ids, kids)
}
length(ids)
}
roots <- setdiff(unique(tmf$inf), unique(tmf$sus))
sizes <- vapply(roots, tree_size, integer(1))
keep <- head(roots[order(sizes)][sort(sizes) >= 3], 3)
if (length(keep) < 1) keep <- head(roots[order(sizes)], 3)
lineage <- keep
repeat {
kids <- tmf$sus[tmf$inf %in% lineage]
if (all(kids %in% lineage)) break
lineage <- union(lineage, kids)
}
tm_sub <- tmf[tmf$inf %in% lineage, ]
class(tm_sub) <- class(tm1)
plot(tm_sub)found multiple trees, returning a list of 3phylo objects