24 Working with Nodal Attributes in Network Models
Our examples so far were all built on a set of nodes that were homogeneous, except for infectious status. In this tutorial, we introduce two groups that can systematically differ in their behavior and their epidemic parameters (for example, the average duration of infection). We motivate our example with a heterosexual network in which the groups represent women and men.
A time step is arbitrary; it takes whatever unit we choose when we set durations and rates. We treat a step as a week in this tutorial, whereas the SIS tutorial treated it as a day.
Download the R script to follow along with this tutorial here.
24.1 Getting Started
To get started, load the EpiModel library.
We also recommend that you clear your environment to avoid any leftover objects from the last tutorials or labs.
24.2 Network Model
For the network model parameterization, we’ll fit a two-group model with a differential degree distribution by group. We will do so using the special group attribute.
In core EpiModel, the group attribute has a special role for these built-in models that we are exploring in this course. It allows for easy parameterization of heterogeneous epidemic parameters (e.g., group-specific recovery rates). This functionality requires that group takes only two values, which must be 1 and 2. In our example, group 1 will represent women and group 2 men.
The group attribute makes life easier in this specific circumstance. In future examples, we can, and will, use other nodal attributes that can take more values (e.g. age or race) and model their impacts on network structure specifically. (Epidemic parameters can also be stratified by an arbitrary number of attributes with arbitrary values; however, this requires some additional programming of the epidemic modules in EpiModel. This is covered in the NME II workshop.)
24.2.1 Initialization
First we set up the network. The number of nodes in each group will be 250:
Network attributes:
vertices = 500
directed = FALSE
hyper = FALSE
loops = FALSE
multiple = FALSE
bipartite = FALSE
total edges= 0
missing edges= 0
non-missing edges= 0
Vertex attribute names:
vertex.names
No edge attributesWe next define our nodal attribute group, and then set it on the network. We can print out the network again to see that it has been added to the list of vertex attributes. Remember, to access the special functionality associated with the group attribute, its values must be 1 and 2 only.
Code
Network attributes:
vertices = 500
directed = FALSE
hyper = FALSE
loops = FALSE
multiple = FALSE
bipartite = FALSE
total edges= 0
missing edges= 0
non-missing edges= 0
Vertex attribute names:
group vertex.names
No edge attributesWe can use get_vertex_attribute to extract the same attribute from the network object:
[1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
[38] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
[75] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
[112] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
[149] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
[186] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
[223] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2
[260] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
[297] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
[334] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
[371] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
[408] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
[445] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
[482] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 224.2.2 Model specification
We are going to specify:
- a constraint that all ties must be cross-sex
- the expected total number of ties
- the expected proportion of each sex that has concurrent ties at any point in time.
Before writing any code, it is worth working out what our own assumptions have already decided for us.
Every tie has two ends. Count them.
- In this model every tie is cross-sex, so every tie puts exactly one end on a woman and one on a man. The women’s tie-ends and the men’s tie-ends are therefore the same number, necessarily.
- A group’s tie-ends divided by its size is its mean degree. Our two groups are the same size.
- So the two groups have the same mean degree.
With the numbers we are about to choose: 165 ties, each with one end on a woman, gives women 165 tie-ends across 250 women, a mean degree of \(165/250 = 0.66\). The men’s arithmetic is identical, because it is the same 165 ties counted from the other end.
This is not a modeling choice we get to make. It is arithmetic, forced by the assumption that all ties are cross-sex plus the fact that the groups are the same size. Change either of those and it stops holding, which is exactly what Lab 2 explores. If you have come from a compartmental model, this is the first time a modeling assumption has silently taken a parameter away from you, and it will not be the last.
We therefore set a single overall mean degree of 0.66, rather than one per group. (The SIS tutorial used 0.7; the exact value is arbitrary in both cases, and nothing here depends on the difference.)
Now consider the proportion of each group that has concurrent ties at any point in time. Is there a similar constraint on how these two numbers relate to each other?
There is not. Consider a population of 4 women and 4 men. Each woman has one partner, and for all four of them it is man #1: the women have 0% concurrency while the men have 25%. Now suppose the first two women partner with man #1 and the other two with man #2: the men have 50% concurrency and the women still have 0%. Mean degree imposes some overall limits (if it exceeds 1, someone must have concurrent partners), and there are constraints at the extremes, but within those the two values move largely independently.
So we specify the proportion of each group with concurrent partners: 5% for women and 15% for men. These values are stylized rather than drawn from a specific survey, but the direction is consistent with egocentric partnership data, in which reported concurrency is typically higher among men.
As you progress into more complex network models, it is helpful to practice thinking through the logical relationships among your terms and their associated statistics carefully. This helps to ensure that you are including the right set of terms for your goal, and not over- or under-specifying the model. We dive into this more deeply in NME-II.
24.2.3 Formation model
Next, we specify the terms in our formation model. As usual, we want an edges term to model the overall mean degree.
The nodematch term should look familiar: you met it in the Module 2 lecture and fit it in the statnetWeb lab, where it modeled homophily by grade and by race. It models assortative or disassortative mixing across an attribute. Assortative mixing means ties form preferentially between nodes that share an attribute value (the same risk group or age group, say); disassortative mixing means ties form preferentially between nodes with different values. This is the first time we write it in R and hand it to EpiModel, and we are using it in the disassortative direction: our assumption is that all ties are cross-sex, which is purely disassortative on sex.
The concurrent term counts the nodes with two or more ongoing partners at a given point in time, the same definition used in the SIS tutorial. It takes an optional by argument naming a nodal attribute to disaggregate the count on, and a levels argument controlling which values of by get their own statistic. Here levels = NULL means “all levels”, which is what we want: one statistic per group. NULL is already this term’s default, so concurrent(by = "group") would be equivalent, and that is the shorter form we use in the netdx call below.
Putting this all together, we get:
As in the SIS tutorial, target.stats is matched positionally to the terms in formation, so we write it one term per line:
- 1
-
edges, as a function of mean degree: \(0.66 \times 500 / 2 = 165\). - 2
-
nodematch("group"). Zero, because purely disassortative mixing means no edges between two people with the same value ofgroup. This one target statistic is what makes every tie cross-sex. - 3
-
concurrent.group1: the expected number of women with concurrent partners, \(0.05 \times 250 = 12.5\). - 4
-
concurrent.group2: the expected number of men, \(0.15 \times 250 = 37.5\).
[1] 165.0 0.0 12.5 37.5Note that it is perfectly fine for our target values for concurrent to not be whole numbers, even though the number of people with concurrent partners in any actual network must be one. The reason is worth stating precisely, because it says what a target statistic is.
An ERGM does not describe a single network. It describes a probability distribution over networks: the random graph from Module 2. Our target statistics are the expected values of the statistics under that distribution, taken across all the networks the model could produce. Any one realization of that random graph has a whole number of nodes with concurrent partners. The expectation across realizations has no reason to be whole, in the same ordinary sense that the average household holds 2.4 children while no particular household does.
The diagnostics below estimate that expectation by averaging over many simulated networks, and over the time steps within each one. That averaging is how we measure the expectation, not why it is fractional: a cross-sectional ERGM with no time dimension at all has fractional target statistics for exactly the same reason.
24.2.4 Dissolution model
The dissolution model is parameterized the same way as the prior example, but with a shorter mean partnership duration of 20 steps. Shorter partnerships turn the network over faster, which is what lets an SIR epidemic reach a large share of the population within the simulated window. It also puts us in the regime the SIS tutorial warned about, where the edges dissolution approximation starts to show some bias, and we will see that in the diagnostics below:
24.2.5 Estimation
The netest function is used for network model fitting.
For the dynamic diagnostics, we simulate from the model fit. We’ll track not only the concurrent statistics but also the low end of the degree distribution, degrees 0 through 3, by group. Note that unlike the SIS tutorial, this model puts no hard cap on degree, so a few nodes can have 4 or more partners; degree(0:3) does not display those, though at a mean degree of 0.66 they are rare:
Code
Network Diagnostics
-----------------------
- Simulating 25 networks
- Calculating formation statisticsEpiModel Network Diagnostics
=======================
Diagnostic Method: Dynamic
Simulations: 25
Time Steps per Sim: 500
Formation Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means)
edges 165.0 163.254 -1.058 0.512 -3.408 3.086
concurrent.group1 12.5 13.198 5.587 0.182 3.831 0.932
concurrent.group2 37.5 36.798 -1.872 0.178 -3.952 1.118
deg0.group.1 NA 106.842 NA 0.284 NA 1.937
deg1.group.1 NA 129.960 NA 0.271 NA 1.684
deg2.group.1 NA 8.190 NA 0.087 NA 0.534
deg3.group.1 NA 3.548 NA 0.056 NA 0.337
deg0.group.2 NA 131.903 NA 0.315 NA 1.895
deg1.group.2 NA 81.299 NA 0.260 NA 1.306
deg2.group.2 NA 29.640 NA 0.147 NA 0.941
deg3.group.2 NA 6.096 NA 0.067 NA 0.377
SD(Statistic)
edges 11.091
concurrent.group1 3.677
concurrent.group2 5.046
deg0.group.1 7.883
deg1.group.1 7.947
deg2.group.1 2.825
deg3.group.1 1.894
deg0.group.2 7.358
deg1.group.2 7.549
deg2.group.2 4.884
deg3.group.2 2.361
Duration Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 20 20.097 0.486 0.074 1.317 0.58 1.565
Dissolution Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.05 0.05 -0.275 0 -0.926 0.001 0.017The edges statistic comes in within a couple of percent of target, while the two concurrent terms show the larger deviations. That split reflects how the approximation works: the edges dissolution approximation analytically corrects the coefficient of each term in the dissolution model, which here is edges only, so edges is the single term it adjusts (Module 5 shows a case with two dissolution terms, where two coefficients get corrected). The dyad-dependent concurrent terms are not in the dissolution model, so they stay unadjusted, and note that they miss in both directions, one group’s concurrency landing a little above target and the other’s a little below, rather than being pulled uniformly one way. That two-sided error is the signature of the unadjusted terms, and it grows as partnership duration gets shorter: the SIS tutorial used a duration of 50 steps and this one uses 20, which is why the bias is visible here and was not there. This is confirmed when we plot the diagnostics:
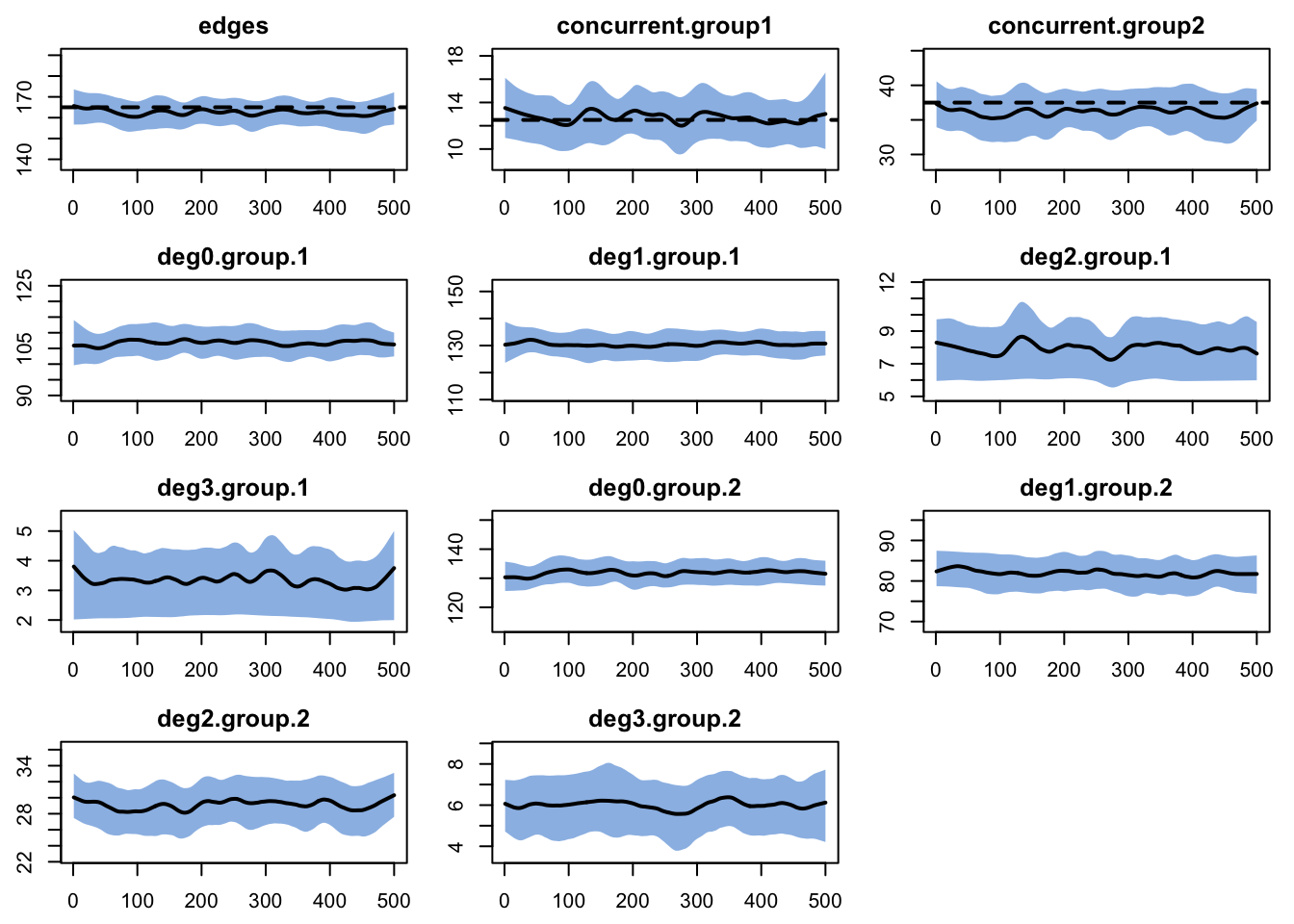
One might want to do a full tergm estimation instead of the approximation. But it is important to consider your goals, and both the relative and absolute magnitude of the bias here. For our purposes, this fit is good enough to move forward.
24.3 Epidemic Model
For the disease simulation, we will simulate an SIR epidemic in a closed population.
24.3.1 Parameterization
The epidemic model parameters are below. We set the per-act infection probability and the recovery rate equal across the two groups, so that any difference in outcome is attributable to the group-specific degree distributions rather than to biology.
Note that the group-specific infection probabilities govern the per-act risk of acquisition for persons in that group, whatever the group of the infecting partner: the parameter is keyed to the susceptible partner’s group, not the infected partner’s. In this model every tie is cross-sex, so in practice inf.prob describes transmission from men to women and inf.prob.g2 from women to men. That coincidence is a property of this network, not of the parameter.
A recovery rate of 0.02 implies a mean infectious duration of \(1/0.02 = 50\) weeks, long relative to the 20-week mean partnership duration, so an infected person will typically have several partnerships while infectious.
Code
- 1
- Per-act acquisition probability for group 1 (women).
- 2
-
Per-act acquisition probability for group 2 (men). The
.g2suffix is the convention throughout: an unsuffixed parameter is group 1, and.g2is group 2. - 3
-
Acts per partnership per time step. The SIS tutorial used 2; here we use 1. This is
param.net’s default, but we state it explicitly rather than let it be silently assumed. - 4
- Recovery rate for group 1, implying a mean infectious duration of 50 weeks.
- 5
- Recovery rate for group 2.
At the outset, 10 people in each group will be infected. For an SIR epidemic simulation, it is necessary to specify the number recovered in each group, here starting at 0.
For control settings, we will simulate 5 epidemics over 500 time steps each. Here we use the multi-core functionality in our controls.
24.3.2 Simulation
The model is simulated by inputting the fitted network model, the epidemic parameters, the initial conditions, and control settings.
Printing the model object shows the inputs and outputs from the simulation object. In particular, take note of the Variables listing; the epidemic outputs for group 1 are listed without a suffix whereas the outputs for group 2 are listed with a .g2 suffix.
EpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type: SIR
No. simulations: 5
No. time steps: 500
No. NW groups: 2
Fixed Parameters
---------------------------
inf.prob = 0.3
act.rate = 1
rec.rate = 0.02
inf.prob.g2 = 0.3
rec.rate.g2 = 0.02
groups = 2
Model Output
-----------------------
Variables: s.num i.num r.num num s.num.g2 i.num.g2
r.num.g2 num.g2 si.flow si.flow.g2 ir.flow ir.flow.g2
Networks: sim1 ... sim5
Transmissions: sim1 ... sim5
Formation Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means)
edges 165.0 161.456 -2.148 0.985 -3.599 2.377
nodematch.group 0.0 0.000 NaN NaN NaN 0.000
concurrent.group1 12.5 12.892 3.139 0.320 1.227 0.811
concurrent.group2 37.5 35.930 -4.188 0.393 -3.998 0.872
SD(Statistic)
edges 10.563
nodematch.group 0.000
concurrent.group1 3.329
concurrent.group2 4.938
Duration Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 20 19.808 -0.959 0.146 -1.312 0.555 1.474
Dissolution Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.05 0.05 0.573 0 0.833 0.001 0.01724.3.3 Analysis
Similar to the first tutorial, plotting the netsim object shows the prevalence for each disease state in the model over time.
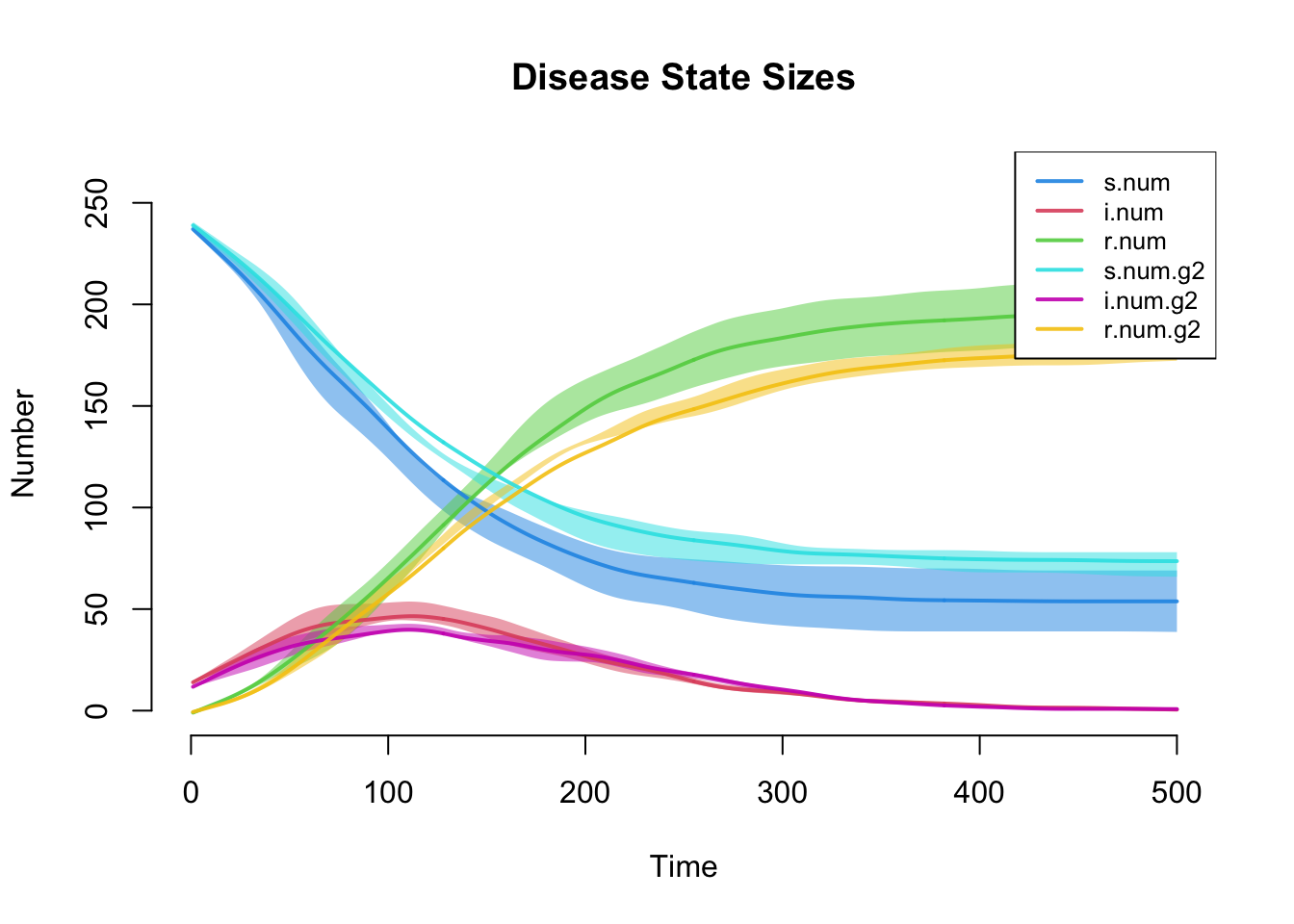
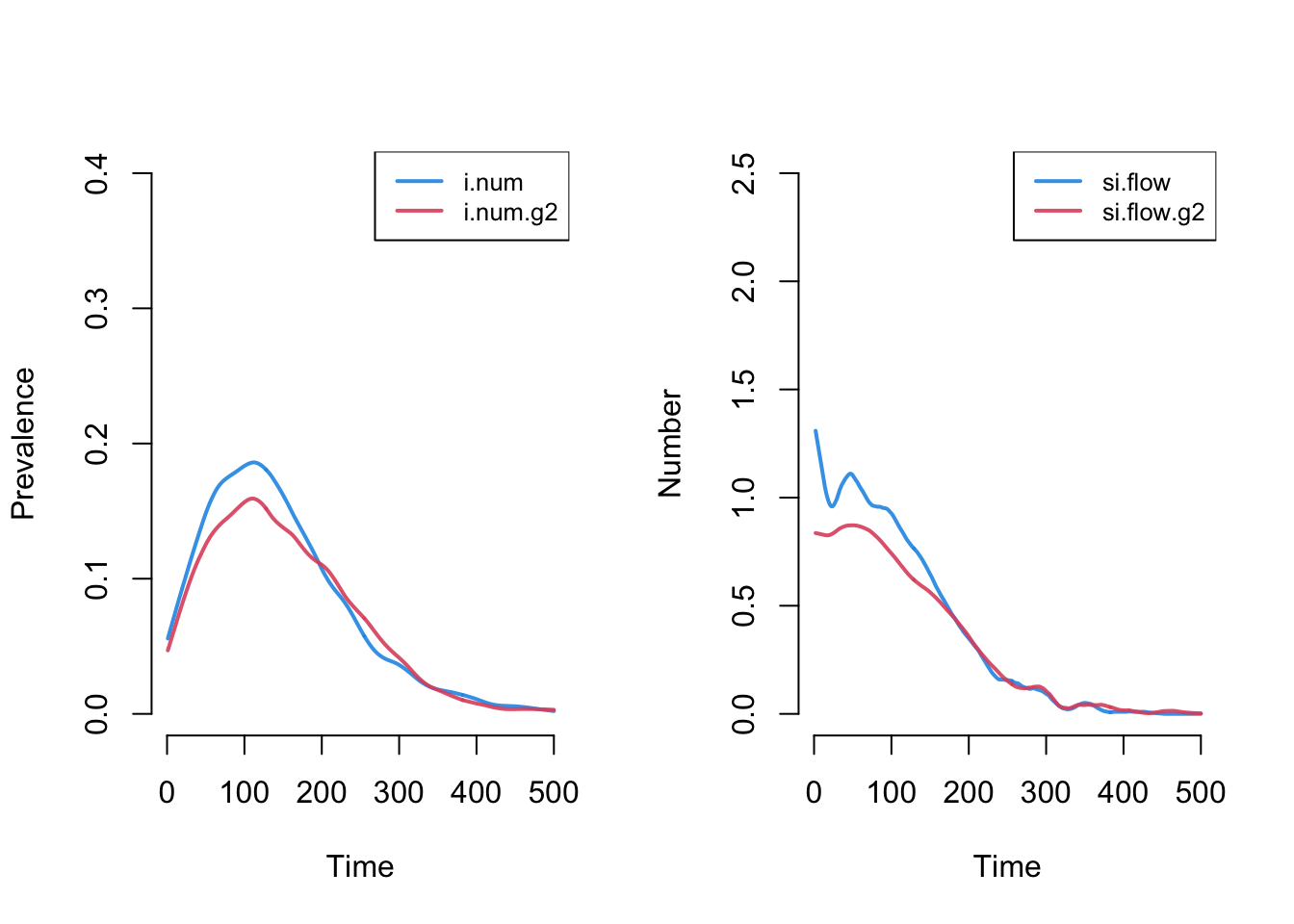
The plot below more clearly shows prevalence and incidence by group. Women (group 1) have a slightly higher prevalence of disease during the main epidemic period, and more women than men end up in the recovered state.
The reason is worth working through. Men and women have the same mean degree, by the tie-end argument above, but men have a higher prevalence of concurrency. Concurrency raises a person’s onward transmission without raising their own acquisition: having two partners at once does not make you more likely to be infected, but it does make you more likely to pass an infection on once you are. So men’s concurrency is a risk that men export rather than absorb, and because every tie is cross-sex, the only people it can be exported to are women.
Note how much this argument depends on the structure we assumed. It needs mean degree to be equal, which our mixing assumption forced, and it needs women’s partners to be men, which it also forced. Lab 2 asks what happens when you relax the second one.
Code
We can also plot the color-coded static network at various time points during the simulation, as in the last tutorial. This helps visualize how a woman’s (circle) risk depends on her male partners’ (square) other connections.
Code

We can add new variables to an existing model object with mutate_epi, which takes inspiration from the mutate functions in the tidyverse. Here we express incidence as an annualized rate per 100 person-years at risk. The time step is a week, so we multiply the per-step incidence (si.flow / s.num) by 52 to annualize it and by 100 to state it per 100 people at risk. This is a change of units, not a standardization: we are not adjusting for the distribution of any confounder, only re-expressing the same rate on an annual, per-100 scale. The mutate_epi function takes a netsim object and adds a new variable.
Printing out the object, you can see that these two new variables are in the variable list.
EpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type: SIR
No. simulations: 5
No. time steps: 500
No. NW groups: 2
Fixed Parameters
---------------------------
inf.prob = 0.3
act.rate = 1
rec.rate = 0.02
inf.prob.g2 = 0.3
rec.rate.g2 = 0.02
groups = 2
Model Output
-----------------------
Variables: s.num i.num r.num num s.num.g2 i.num.g2
r.num.g2 num.g2 si.flow si.flow.g2 ir.flow ir.flow.g2 ir.g1
ir.g2
Networks: sim1 ... sim5
Transmissions: sim1 ... sim5
Formation Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means)
edges 165.0 161.456 -2.148 0.985 -3.599 2.377
nodematch.group 0.0 0.000 NaN NaN NaN 0.000
concurrent.group1 12.5 12.892 3.139 0.320 1.227 0.811
concurrent.group2 37.5 35.930 -4.188 0.393 -3.998 0.872
SD(Statistic)
edges 10.563
nodematch.group 0.000
concurrent.group1 3.329
concurrent.group2 4.938
Duration Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 20 19.808 -0.959 0.146 -1.312 0.555 1.474
Dissolution Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.05 0.05 0.573 0 0.833 0.001 0.017After we add the new variable, we can plot and analyze it like any other built-in variable.
Code
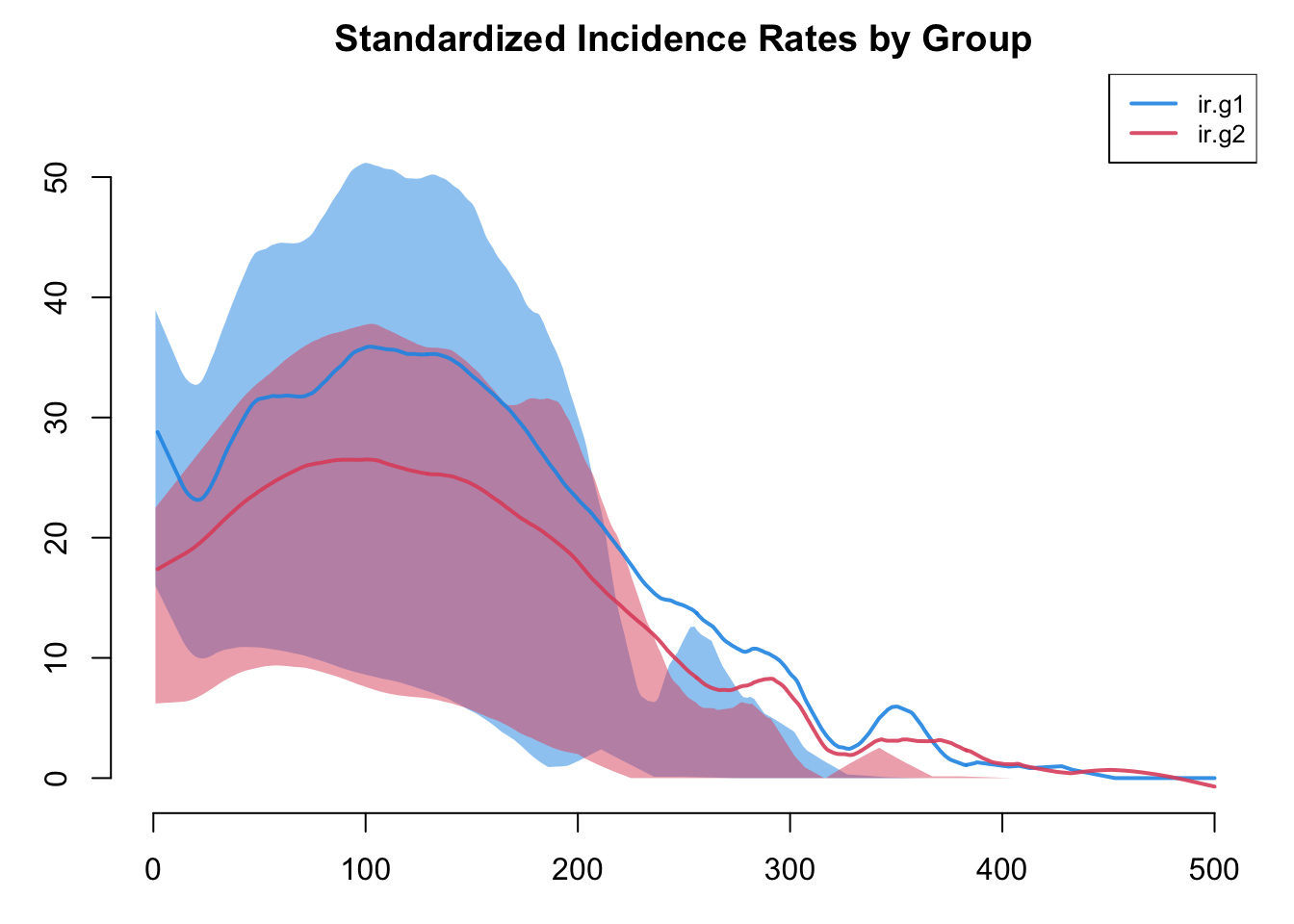
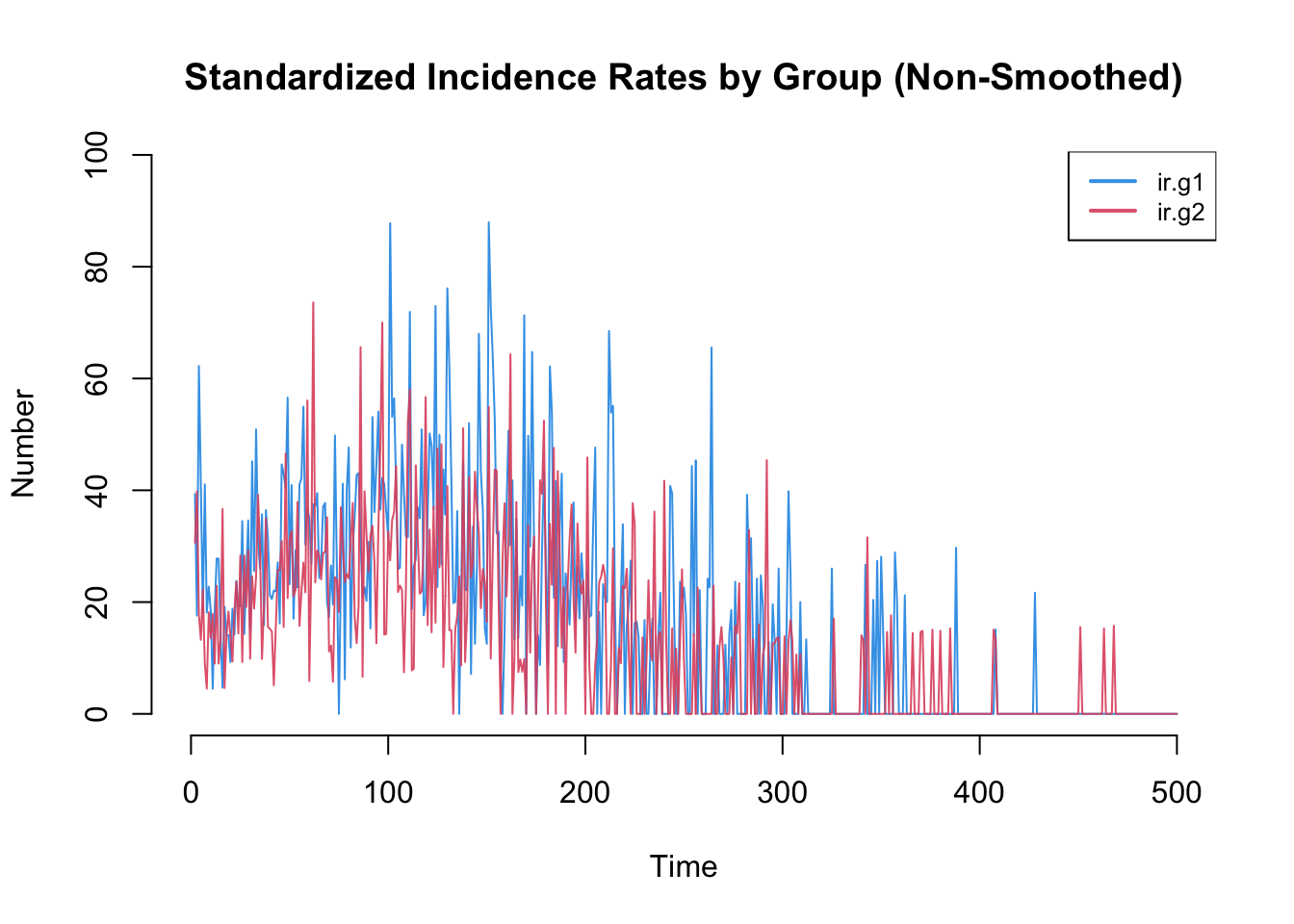
Incidence rates in particular are often highly variable. The plot by default will use a loess smoother so some of this variability may be lost. Therefore, it is worth inspecting the non-smoothed plots too.
Code
As another exercise, let’s extract the data into a data frame and then calculate some other relevant summary statistics. By default as.data.frame will extract the individual simulation data in each row, but by specifying out = "mean" we can see the time specific averages for all the variables across simulations. The variables in this data frame include both the original variables from netsim plus the two new ones we added with mutate_epi.
time s.num i.num r.num num s.num.g2 i.num.g2 r.num.g2 num.g2 si.flow
1 1 240.0 10.0 0.0 250 240.0 10.0 0.0 250 NaN
2 2 238.6 11.4 0.0 250 238.6 11.2 0.2 250 1.4
3 3 237.6 12.0 0.4 250 237.4 12.2 0.4 250 1.0
4 4 235.0 14.2 0.8 250 236.6 12.8 0.6 250 2.6
5 5 233.2 16.0 0.8 250 236.0 13.0 1.0 250 1.8
6 6 232.8 16.2 1.0 250 235.4 13.4 1.2 250 0.4
7 7 232.0 17.0 1.0 250 234.6 13.6 1.8 250 0.8
8 8 231.6 17.4 1.0 250 233.8 14.4 1.8 250 0.4
9 9 231.0 17.8 1.2 250 233.6 13.8 2.6 250 0.6
10 10 230.2 17.8 2.0 250 233.0 13.8 3.2 250 0.8
si.flow.g2 ir.flow ir.flow.g2 ir.g1 ir.g2
1 NaN NaN NaN NaN NaN
2 1.4 0.0 0.2 30.643549 30.681512
3 1.2 0.4 0.2 21.978116 26.652247
4 0.8 0.4 0.2 57.647947 17.534305
5 0.6 0.0 0.4 40.368360 13.450258
6 0.6 0.2 0.2 8.870299 13.183465
7 0.8 0.0 0.6 17.835167 17.740598
8 0.8 0.0 0.0 9.122807 17.778427
9 0.2 0.2 0.8 13.547015 4.502165
10 0.6 0.8 0.6 18.126448 13.487256One useful summary for an SIR epidemic is the cumulative number of infections, the total count of new infections over the whole simulation. Summing the mean si.flow gives that count in each group across simulations, out of 250 people per group. We use na.rm = TRUE to drop the first row, where the flow is undefined. Note that this is a count of incident infections, not the cumulative incidence (the proportion of the at-risk group that became infected); dividing the count by the number susceptible at the outset (240 per group, since 10 of the 250 begin infected) would give that risk. Lab 2 asks you to compare against these two numbers, so note them down.
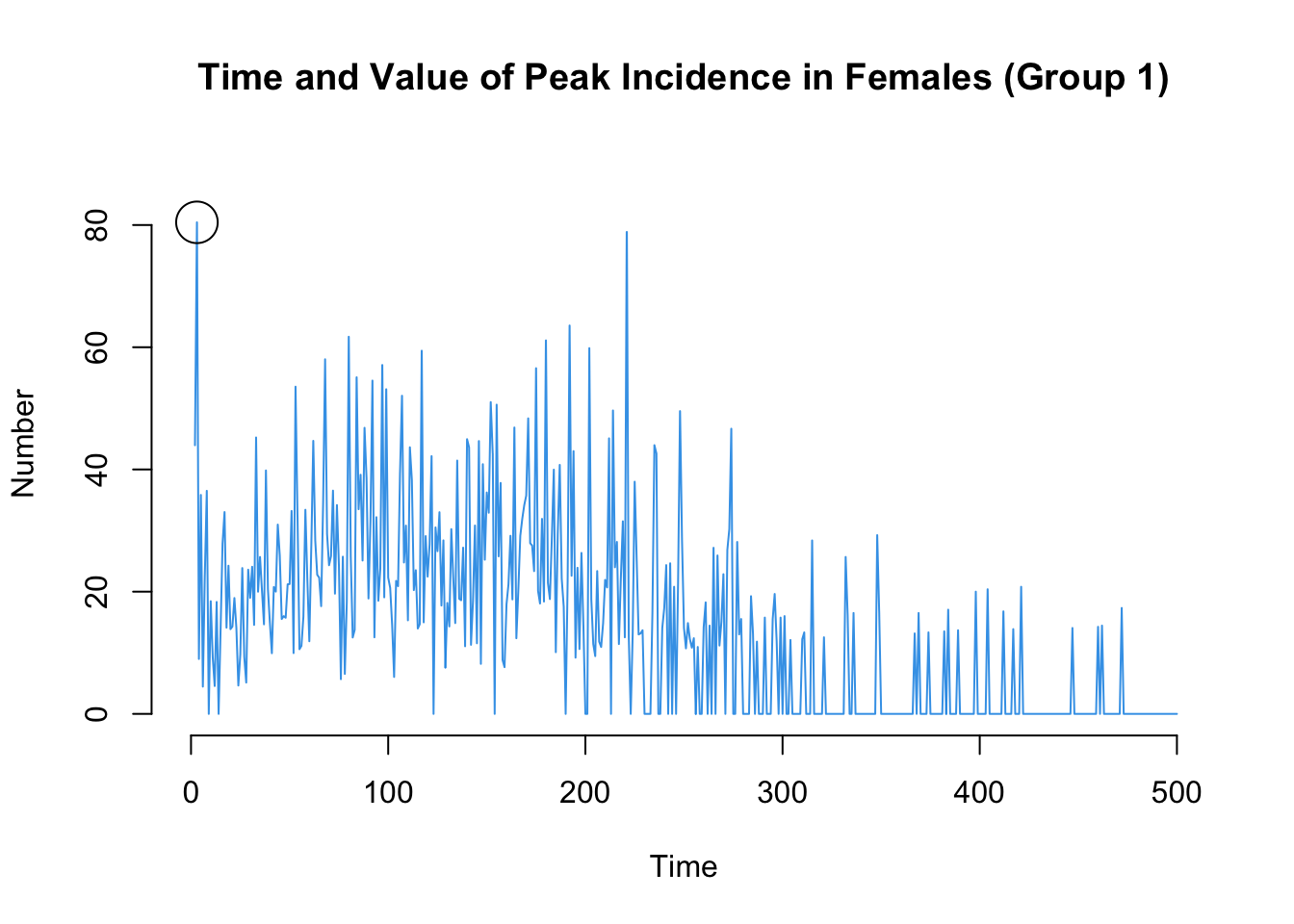
Here are two more that may be of interest: the time point of the peak annualized incidence rate, and also the value of the incidence rate at that time point.
Code
The transmission matrix again shows the individual transmission events that occurred at each time step, along with information about the infecting partner that may be relevant.
# A tibble: 10 × 8
# Groups: at, sus [10]
at sus inf network infDur transProb actRate finalProb
<int> <int> <int> <int> <dbl> <dbl> <dbl> <dbl>
1 2 58 443 1 10 0.3 1 0.3
2 2 180 443 1 10 0.3 1 0.3
3 2 452 26 1 61 0.3 1 0.3
4 3 11 489 1 60 0.3 1 0.3
5 3 225 489 1 60 0.3 1 0.3
6 4 3 379 1 84 0.3 1 0.3
7 4 492 142 1 56 0.3 1 0.3
8 5 13 492 1 1 0.3 1 0.3
9 6 300 15 1 53 0.3 1 0.3
10 7 190 346 1 5 0.3 1 0.3The infDur column shows the duration of infection (in time steps) of the infecting partner at the point of transmission. For example, an event with an infDur of 1 means that the infecting partner had just become infected themselves in the prior time step. The code below computes the proportion of transmission events for which the infecting partner had just become infected (infDur of 1).
One can also arrange these transmission chains as a tree for easier visualization, using the phylo class from the ape package. First, we convert the transmission matrix into a phylo object that can then be analyzed and visualized with ape’s tools. In this transmission matrix, multiple trees are found because we seeded the infection with 20 people as initial conditions. Only a seed that went on to infect at least one other person becomes the root of a tree, so the number of trees is at most 20 and is usually somewhat fewer, since some seeds recover before passing the infection on.
The phylo object here is a container borrowed from phylogenetics, but what it holds is the model’s exact, known transmission tree: get_transmat recorded who infected whom at every step, so this tree is observed ground truth with no estimation involved.
A molecular phylogeny is a different thing. In a real outbreak the transmission tree is unobserved; a phylogeny is estimated from pathogen genetic sequences, reconstructs the ancestry of those sequences (which only approximates who infected whom), and carries real statistical uncertainty. Reusing the ape phylo class and its plots does not make our simulated tree a phylogenetic inference; it just borrows phylogenetics’ drawing tools. The point of a simulation like this is precisely that we hold the true tree, which is what lets it serve as a benchmark for methods that must estimate the tree from sequence data.
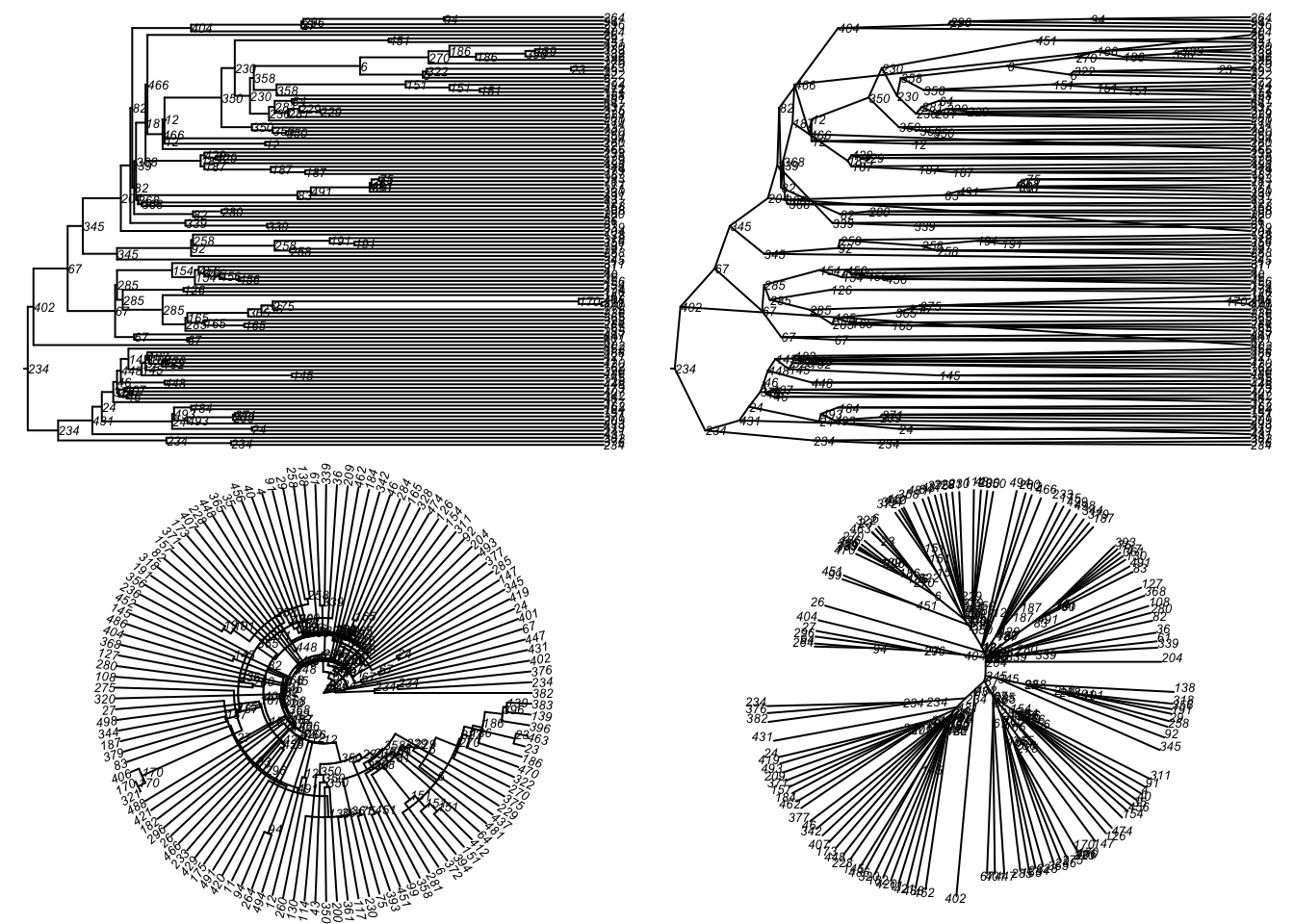
Here is a standard phylogram of the first four trees. Some initial seeds lead to many downstream infections whereas others do not. Whether that reflects the seeded nodes’ network position, their biology or behavior, or chance alone is a question this model could be used to answer.
Code
par(mfrow = c(2, 2), mar = c(0, 0, 0, 0))
plot(tmPhylo[[1]], show.node.label = TRUE, root.edge = TRUE, cex = 0.5)
plot(tmPhylo[[2]], show.node.label = TRUE, root.edge = TRUE, cex = 0.5)
plot(tmPhylo[[3]], show.node.label = TRUE, root.edge = TRUE, cex = 0.5)
plot(tmPhylo[[4]], show.node.label = TRUE, root.edge = TRUE, cex = 0.5)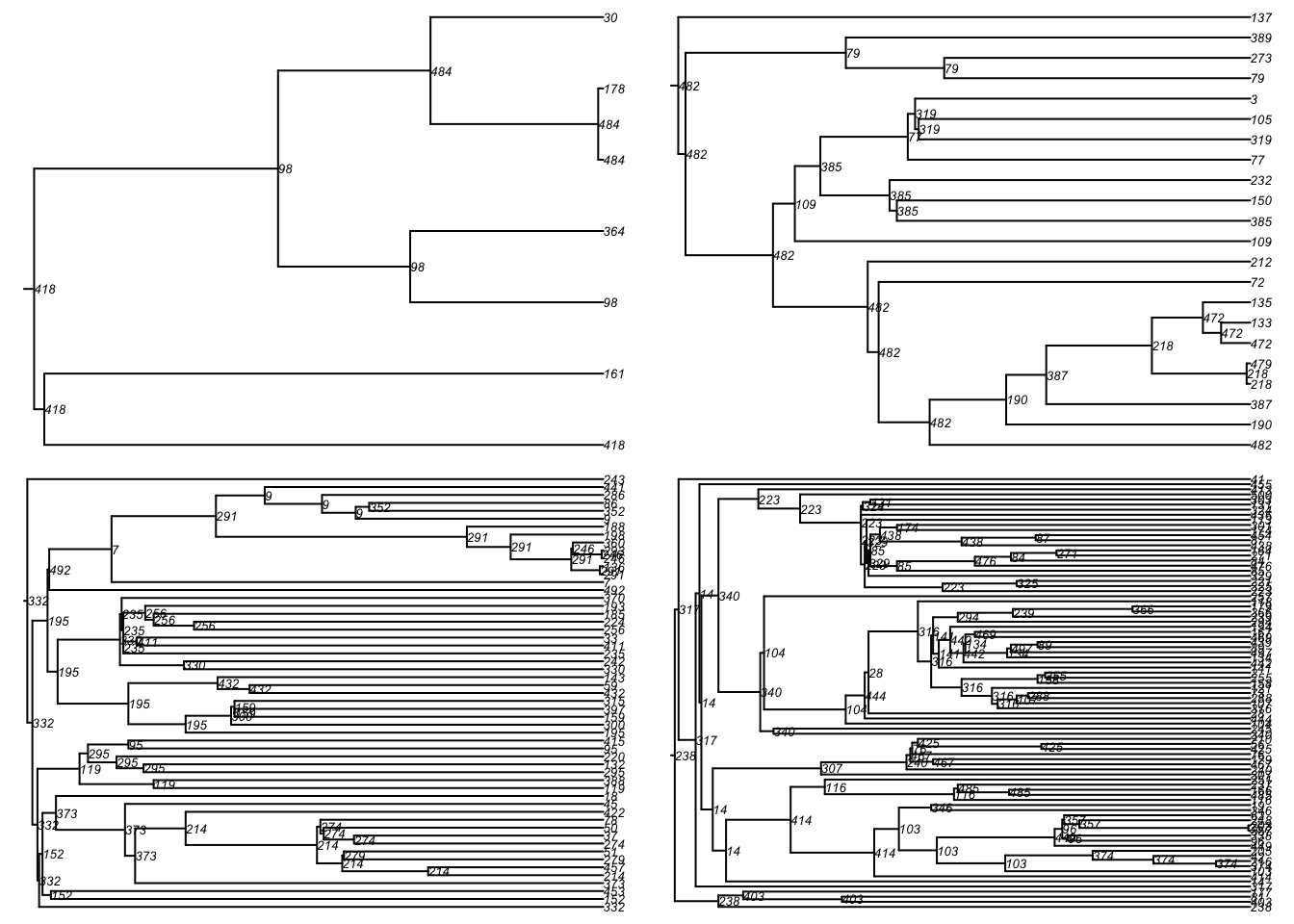
In phylo objects with multiple trees, each tree is stored with a named object. Note that these names correspond to the ID numbers in the transmission matrix itself. We can extract one tree like so:
[1] "seed_443" "seed_26" "seed_489" "seed_379" "seed_142" "seed_15"
[7] "seed_346" "seed_120" "seed_373" "seed_18" "seed_267" "seed_69"
[13] "seed_166"Then we can plot that particular tree as a phylogram and other visual types included in the ape package.
Code
par(mfrow = c(2, 2), mar = c(0, 0, 0, 0))
plot(tmPhylo5, type = "phylogram",
show.node.label = TRUE, root.edge = TRUE, cex = 0.5)
plot(tmPhylo5, type = "cladogram",
show.node.label = TRUE, root.edge = TRUE, cex = 0.5)
plot(tmPhylo5, type = "fan",
show.node.label = TRUE, root.edge = TRUE, cex = 0.5)
plot(tmPhylo5, type = "unrooted",
show.node.label = TRUE, root.edge = TRUE, cex = 0.5)