flowchart LR
in(( )) -->|"arrival<br/>(age 0)"| S
S["<b>S</b><br/>Susceptible"] -->|"infection<br/>(si.flow)"| I["<b>I</b><br/>Infectious"]
S -->|"age-specific<br/>mortality"| out1(( ))
I -->|"age-specific ×<br/>disease multiplier"| out2(( ))
style S fill:#3498db,color:#fff
style I fill:#e74c3c,color:#fff
style in fill:none,stroke:none
style out1 fill:none,stroke:none
style out2 fill:none,stroke:none
SI Model with Age-Specific Vital Dynamics
SI
vital dynamics
aging
beginner
An SI epidemic on a dynamic network with aging, births, and age-specific mortality using US life table data.
Overview
This example demonstrates how to model an SI epidemic on a dynamic network with vital dynamics — aging, births (arrivals), and deaths (departures). In most real-world settings, populations are open: individuals are born, age, and die over the course of an epidemic. These demographic processes shape disease dynamics by continuously introducing new susceptibles (via births) and removing individuals (via death).
The key extension here is age-specific mortality using real US mortality data, combined with disease-induced excess mortality where infected individuals face a higher death rate. The network model also includes age-assortative mixing, so partnerships form preferentially between individuals close in age.
This is the foundational vital dynamics example in the Gallery. The three custom modules (aging, departures, arrivals) are building blocks reused and extended in many other examples.
TipDownload standalone scripts
- model.R — Main simulation script
- module-fx.R — Custom module functions
Model Structure
| Compartment | Label | Description |
|---|---|---|
| Susceptible | S | Not infected; at risk of infection through contact |
| Infectious | I | Infected and capable of transmitting (no recovery in SI) |
Three custom modules implement the demographic processes:
- Aging: Each timestep represents one week. All nodes age by 1/52 years per step.
- Departures (mortality): Age-specific weekly mortality from US life table data. Infected individuals have their rate multiplied by
departure.disease.mult. - Arrivals (births): New susceptible nodes enter at age 0. The arrival rate is calibrated to the mean departure rate for approximate population stability.
Setup
suppressMessages(library(EpiModel))
nsims <- 5
ncores <- 5
nsteps <- 256Vital Dynamics Data
Age-specific mortality rates from US population data, converted from per-100,000-per-year to per-person-per-week probabilities.
ages <- 0:85
# Age-specific mortality rates per 100,000 per year (US)
departure_rate <- c(588.45, 24.8, 11.7, 14.55, 47.85, 88.2, 105.65, 127.2,
154.3, 206.5, 309.3, 495.1, 736.85, 1051.15, 1483.45,
2294.15, 3642.95, 6139.4, 13938.3)
1dep_rate_pw <- departure_rate / 1e5 / 52
2age_spans <- c(1, 4, rep(5, 16), 1)
3dr_vec <- rep(dep_rate_pw, times = age_spans)- 1
- Convert from per-100,000-per-year to per-person-per-week (our timestep unit).
- 2
- Age groups: 1 year for <1, 4 years for 1–4, then 5-year groups through 80–84, and 1 year for 85+.
- 3
- Expand the 19 age-group rates into a vector of 86 yearly rates.
par(mar = c(3, 3, 2, 1), mgp = c(2, 1, 0))
barplot(dr_vec, col = "steelblue", border = NA,
xlab = "Age", ylab = "Weekly Departure Rate",
main = "Age-Specific Mortality Rates (US)")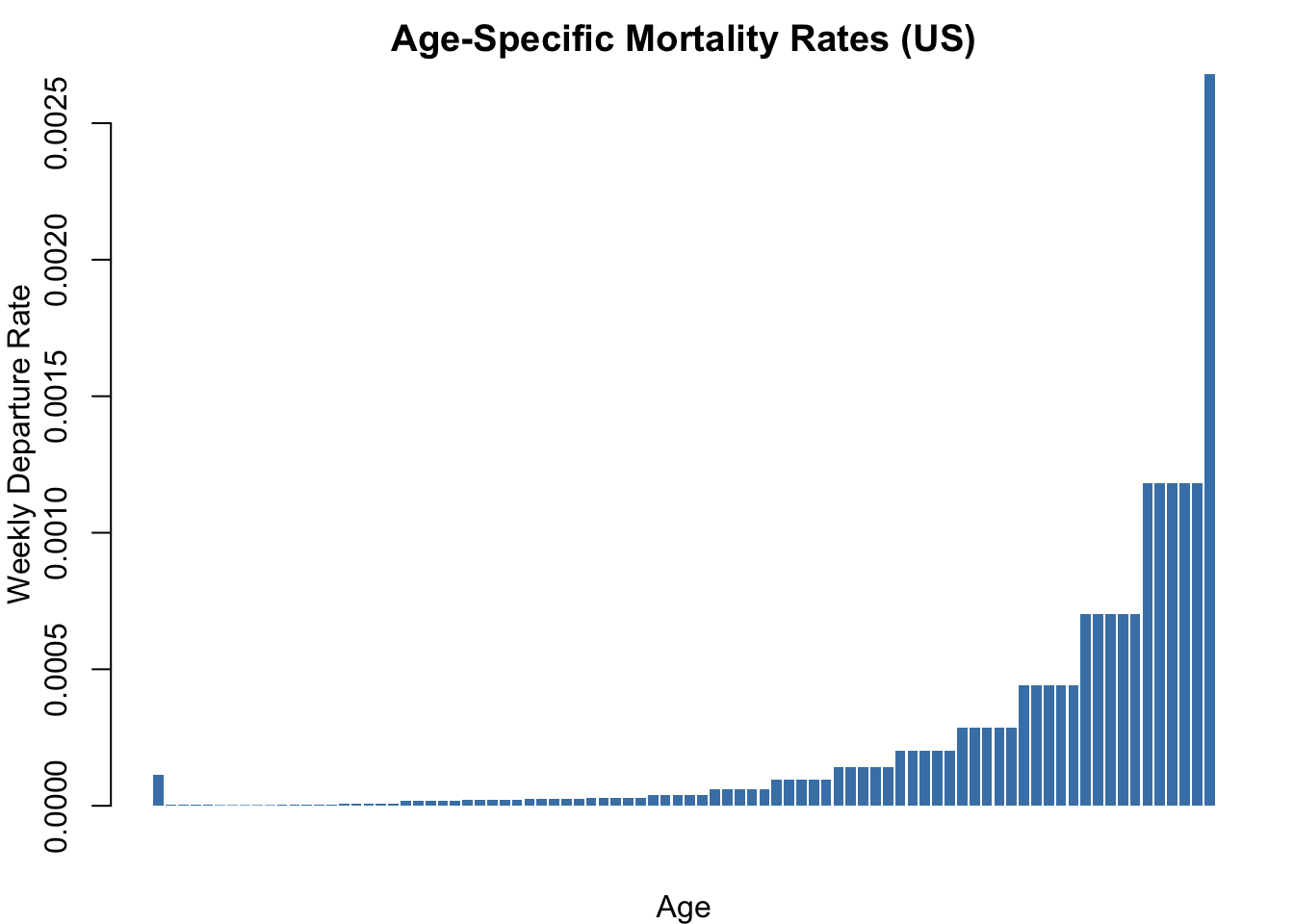
Custom Modules
Aging Module
Increments each node’s age by 1/52 per timestep and records the population mean age.
- 1
- One timestep = one week, so age increments by 1/52 of a year.
- 2
- Track mean population age as an epidemiological summary statistic.
Departure Module
Simulates age-specific mortality with optional disease-induced excess mortality.
dfunc <- function(dat, at) {
active <- get_attr(dat, "active")
exitTime <- get_attr(dat, "exitTime")
age <- get_attr(dat, "age")
status <- get_attr(dat, "status")
dep.rates <- get_param(dat, "departure.rates")
dep.dis.mult <- get_param(dat, "departure.disease.mult")
idsElig <- which(active == 1)
nElig <- length(idsElig)
nDepts <- 0
if (nElig > 0) {
1 whole_ages_of_elig <- pmin(ceiling(age[idsElig]), 86)
departure_rates_of_elig <- dep.rates[whole_ages_of_elig]
2 idsElig.inf <- which(status[idsElig] == "i")
departure_rates_of_elig[idsElig.inf] <-
departure_rates_of_elig[idsElig.inf] * dep.dis.mult
3 vecDepts <- which(rbinom(nElig, 1, departure_rates_of_elig) == 1)
idsDepts <- idsElig[vecDepts]
nDepts <- length(idsDepts)
if (nDepts > 0) {
active[idsDepts] <- 0
exitTime[idsDepts] <- at
}
}
dat <- set_attr(dat, "active", active)
dat <- set_attr(dat, "exitTime", exitTime)
dat <- set_epi(dat, "d.flow", at, nDepts)
return(dat)
}- 1
-
Map continuous age to the 86-element rate vector index.
pmin(..., 86)caps ages 85+ at the final rate. - 2
- Infected individuals face excess mortality: their rate is multiplied by the disease multiplier parameter.
- 3
- Bernoulli trial: each eligible node departs with their age-specific (and disease-adjusted) probability.
Arrival Module
Simulates births as new susceptible nodes entering at age 0.
afunc <- function(dat, at) {
n <- sum(get_attr(dat, "active") == 1)
a.rate <- get_param(dat, "arrival.rate")
nArrivalsExp <- n * a.rate
1 nArrivals <- rpois(1, nArrivalsExp)
if (nArrivals > 0) {
2 dat <- append_core_attr(dat, at, nArrivals)
dat <- append_attr(dat, "status", "s", nArrivals)
dat <- append_attr(dat, "infTime", NA, nArrivals)
3 dat <- append_attr(dat, "age", 0, nArrivals)
}
dat <- set_epi(dat, "a.flow", at, nArrivals)
return(dat)
}- 1
- Number of births is Poisson-distributed, scaled by current population size.
- 2
-
append_core_attr()initializes required node attributes (active status, unique ID, etc.). - 3
- All newborns enter at age 0 as susceptibles.
Network Model
n <- 500
nw <- network_initialize(n)
ageVec <- sample(ages, n, replace = TRUE)
nw <- set_vertex_attribute(nw, "age", ageVec)
# Formation: edges + age-assortative mixing
1formation <- ~edges + absdiff("age")
mean_degree <- 0.8
n_edges <- mean_degree * (n / 2)
avg.abs.age.diff <- 1.5
2target.stats <- c(n_edges, n_edges * avg.abs.age.diff)
# Dissolution: adjust for population turnover
coef.diss <- dissolution_coefs(~offset(edges), duration = 60,
3 d.rate = mean(dr_vec))
coef.diss
est <- netest(nw, formation, target.stats, coef.diss)- 1
-
absdiff("age")controls age-assortative mixing — partners tend to be close in age. - 2
- Target: mean degree of 0.8 with average absolute age difference of 1.5 years within partnerships.
- 3
-
The
d.rateargument adjusts dissolution coefficients for population turnover so observed partnership duration matches the intended 60 weeks.
Dissolution Coefficients
=======================
Dissolution Model: ~offset(edges)
Target Statistics: 60
Crude Coefficient: 4.077537
Mortality/Exit Rate: 0.0002217519
Adjusted Coefficient: 4.104505Network Diagnostics
dx <- netdx(est, nsims = nsims, ncores = ncores, nsteps = nsteps,
nwstats.formula = ~edges + absdiff("age") + isolates + degree(0:5))
Network Diagnostics
-----------------------
- Simulating 5 networks
- Calculating formation statisticsprint(dx)EpiModel Network Diagnostics
=======================
Diagnostic Method: Dynamic
Simulations: 5
Time Steps per Sim: 256
Formation Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 200 194.680 -2.660 2.166 -2.456 3.981 11.402
absdiff.age 300 296.666 -1.111 5.783 -0.577 9.377 28.368
isolates NA 230.510 NA 2.399 NA 5.695 12.771
degree0 NA 230.510 NA 2.399 NA 5.695 12.771
degree1 NA 176.392 NA 1.778 NA 4.471 11.375
degree2 NA 70.998 NA 1.180 NA 1.888 7.961
degree3 NA 17.926 NA 0.452 NA 0.783 3.662
degree4 NA 3.713 NA 0.220 NA 0.308 1.912
degree5 NA 0.423 NA 0.069 NA 0.118 0.643
Duration Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 60 58.585 -2.358 0.703 -2.012 2.082 3.95
Dissolution Diagnostics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.017 0.017 2.571 0 1.718 0.001 0.009plot(dx)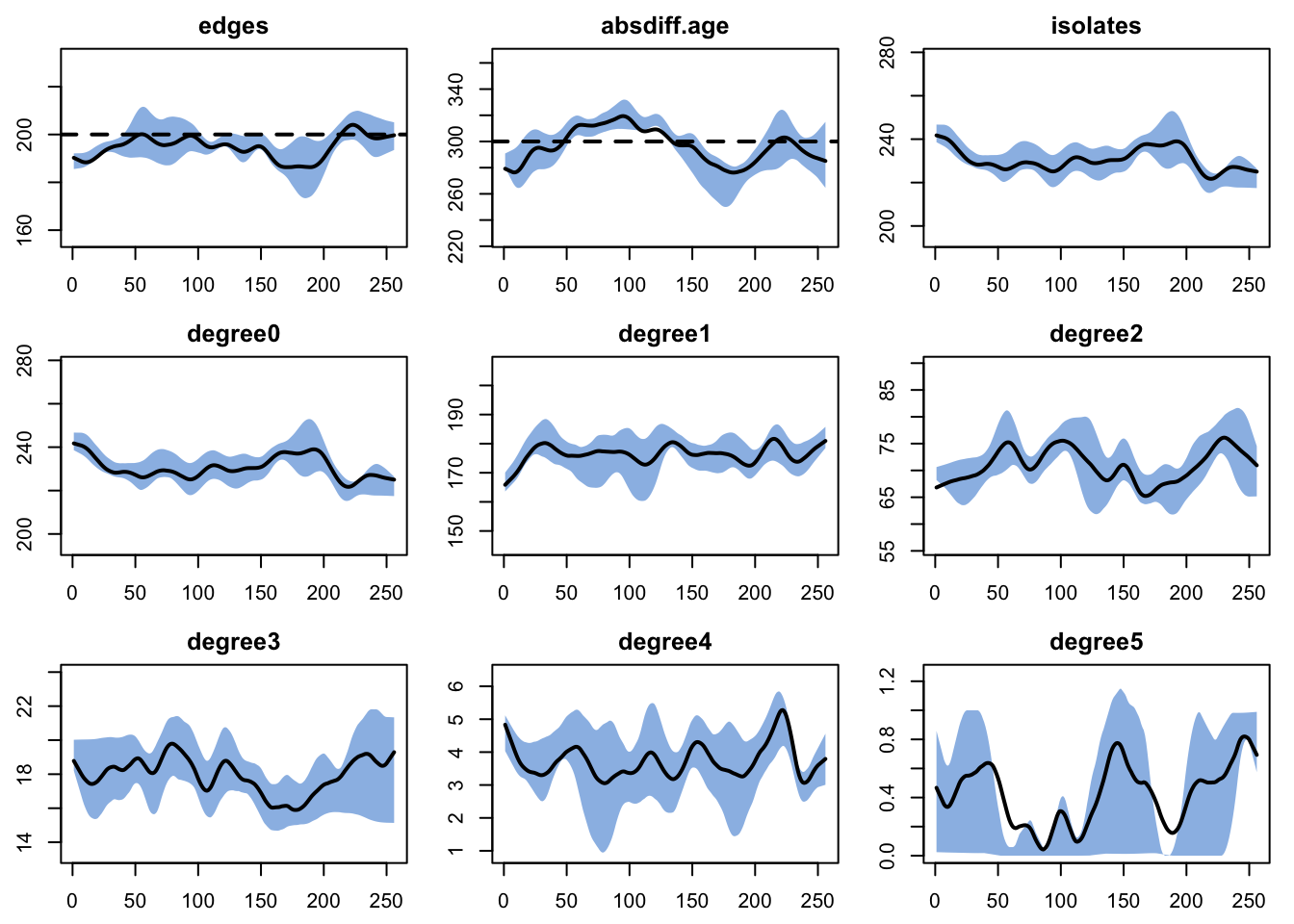
Epidemic Simulation
Scenario 1: No Disease-Induced Mortality (Baseline)
All-cause mortality only. Disease has no effect on survival, so the population size remains stable and prevalence rises toward saturation.
init <- init.net(i.num = 50)
param_base <- param.net(
inf.prob = 0.15,
departure.rates = dr_vec,
1 departure.disease.mult = 1,
arrival.rate = mean(dr_vec)
)
control <- control.net(
2 type = NULL,
nsims = nsims,
ncores = ncores,
nsteps = nsteps,
aging.FUN = aging,
departures.FUN = dfunc,
arrivals.FUN = afunc,
infection.FUN = infection.net,
resim_nets.FUN = resim_nets,
3 resimulate.network = TRUE,
verbose = FALSE
)
sim_base <- netsim(est, param_base, init, control)
print(sim_base)- 1
- Multiplier of 1 = disease has no effect on mortality.
- 2
-
type = NULLmeans all modules are custom (no built-in SIS/SIR logic). - 3
-
Network must be resimulated each step because ages change and the ERGM formation model includes
absdiff("age").
EpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type:
No. simulations: 5
No. time steps: 256
No. NW groups: 1
Fixed Parameters
---------------------------
inf.prob = 0.15
departure.rates = 0.0001131635 4.769231e-06 4.769231e-06 4.769231e-06
4.769231e-06 2.25e-06 2.25e-06 2.25e-06 2.25e-06 2.25e-06 ...
departure.disease.mult = 1
arrival.rate = 0.0002217519
act.rate = 1
groups = 1
Model Functions
-----------------------
initialize.FUN
resim_nets.FUN
summary_nets.FUN
infection.FUN
departures.FUN
arrivals.FUN
nwupdate.FUN
prevalence.FUN
verbose.FUN
aging.FUN
Model Output
-----------------------
Variables: s.num i.num num meanAge si.flow d.flow a.flow
Networks: sim1 ... sim5
Transmissions: sim1 ... sim5
Formation Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 200 200.079 0.039 2.631 0.030 6.601 13.035
absdiff.age 300 288.584 -3.805 5.645 -2.022 20.211 29.951
Duration Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 60 61.287 2.145 0.511 2.52 1.201 2.936
Dissolution Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.017 0.016 -1.673 0 -1.109 0 0.009Scenario 2: Disease-Induced Mortality (50x)
Infected individuals face 50x the baseline age-specific mortality rate — exaggerated for pedagogical clarity.
param_lethal <- param.net(
inf.prob = 0.15,
departure.rates = dr_vec,
departure.disease.mult = 50,
arrival.rate = mean(dr_vec)
)
sim_lethal <- netsim(est, param_lethal, init, control)
print(sim_lethal)EpiModel Simulation
=======================
Model class: netsim
Simulation Summary
-----------------------
Model type:
No. simulations: 5
No. time steps: 256
No. NW groups: 1
Fixed Parameters
---------------------------
inf.prob = 0.15
departure.rates = 0.0001131635 4.769231e-06 4.769231e-06 4.769231e-06
4.769231e-06 2.25e-06 2.25e-06 2.25e-06 2.25e-06 2.25e-06 ...
departure.disease.mult = 50
arrival.rate = 0.0002217519
act.rate = 1
groups = 1
Model Functions
-----------------------
initialize.FUN
resim_nets.FUN
summary_nets.FUN
infection.FUN
departures.FUN
arrivals.FUN
nwupdate.FUN
prevalence.FUN
verbose.FUN
aging.FUN
Model Output
-----------------------
Variables: s.num i.num num meanAge si.flow d.flow a.flow
Networks: sim1 ... sim5
Transmissions: sim1 ... sim5
Formation Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 200 159.886 -20.057 5.66 -7.088 8.029 20.224
absdiff.age 300 245.053 -18.316 8.07 -6.809 9.163 32.691
Duration Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 60 57.488 -4.187 0.644 -3.899 0.824 3.591
Dissolution Statistics
-----------------------
Target Sim Mean Pct Diff Sim SE Z Score SD(Sim Means) SD(Statistic)
edges 0.017 0.019 13.745 0 7.077 0 0.011Analysis
sim_base <- mutate_epi(sim_base, prev = i.num / num)
sim_lethal <- mutate_epi(sim_lethal, prev = i.num / num)Prevalence Comparison
Without disease mortality, SI prevalence saturates near 1 as everyone eventually becomes infected. With lethal disease, mortality removes infected individuals before they can transmit, slowing the epidemic.
par(mfrow = c(1, 1), mar = c(3, 3, 2, 1), mgp = c(2, 1, 0))
plot(sim_base, y = "prev",
main = "Prevalence: Baseline vs. Disease Mortality",
ylab = "Prevalence (I / N)", xlab = "Week",
mean.col = "steelblue", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "steelblue", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE)
plot(sim_lethal, y = "prev",
mean.col = "firebrick", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "firebrick", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE, add = TRUE)
legend("topleft", legend = c("Baseline (mult=1)", "Lethal (mult=50)"),
col = c("steelblue", "firebrick"), lwd = 2, bty = "n")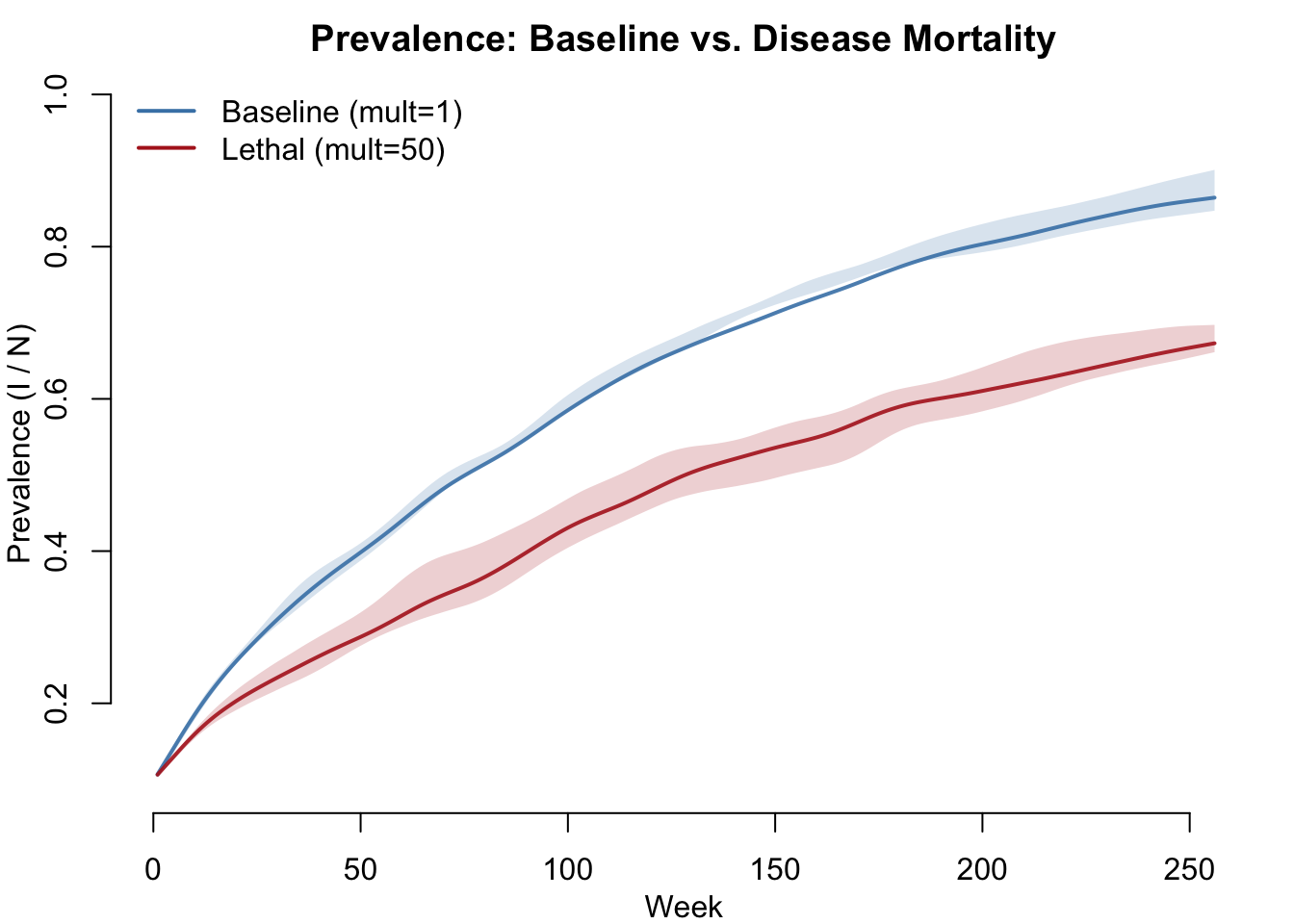
Population Size
Baseline: population stable (arrivals approximately equal departures). Lethal: population declines because disease-induced mortality exceeds the birth rate calibrated to background mortality only.
par(mfrow = c(1, 1), mar = c(3, 3, 2, 1), mgp = c(2, 1, 0))
plot(sim_base, y = "num",
main = "Population Size",
ylab = "Total Population", xlab = "Week",
mean.col = "steelblue", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "steelblue", qnts.alpha = 0.2, qnts.smooth = TRUE,
ylim = c(200, 600), legend = FALSE)
plot(sim_lethal, y = "num",
mean.col = "firebrick", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "firebrick", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE, add = TRUE)
legend("topright", legend = c("Baseline (mult=1)", "Lethal (mult=50)"),
col = c("steelblue", "firebrick"), lwd = 2, bty = "n")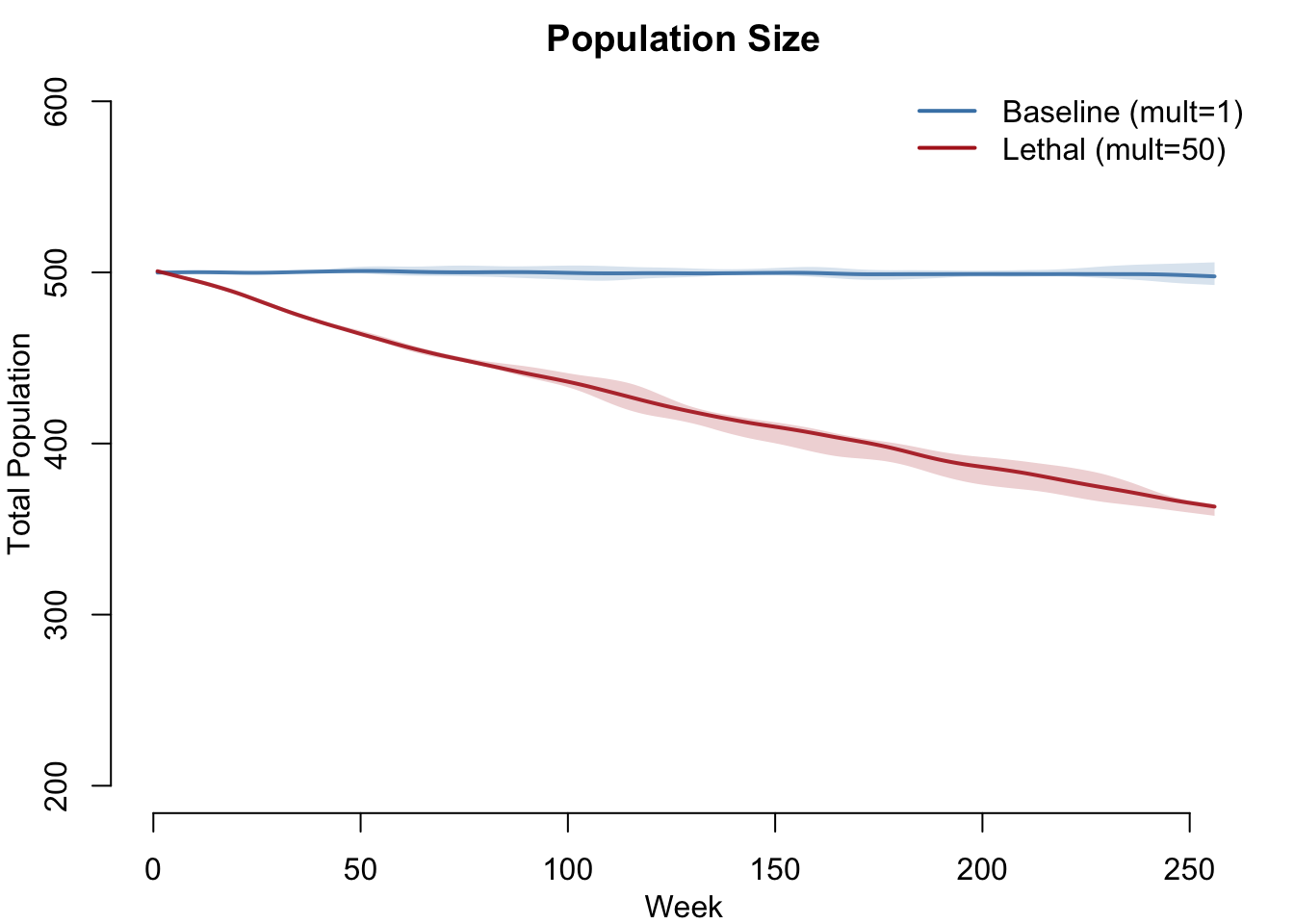
Vital Dynamics Detail (Lethal Scenario)
par(mfrow = c(2, 2), mar = c(3, 3, 2, 1), mgp = c(2, 1, 0))
plot(sim_lethal, y = "meanAge",
main = "Mean Age", ylab = "Years", xlab = "Week",
mean.col = "firebrick", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "firebrick", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE)
plot(sim_lethal, y = "d.flow",
main = "Departures per Week", ylab = "Count", xlab = "Week",
mean.col = "firebrick", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "firebrick", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE)
plot(sim_lethal, y = "a.flow",
main = "Arrivals per Week", ylab = "Count", xlab = "Week",
mean.col = "#27ae60", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "#27ae60", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE)
plot(sim_lethal, y = "s.num",
main = "Susceptible Count", ylab = "Count", xlab = "Week",
mean.col = "steelblue", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "steelblue", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE)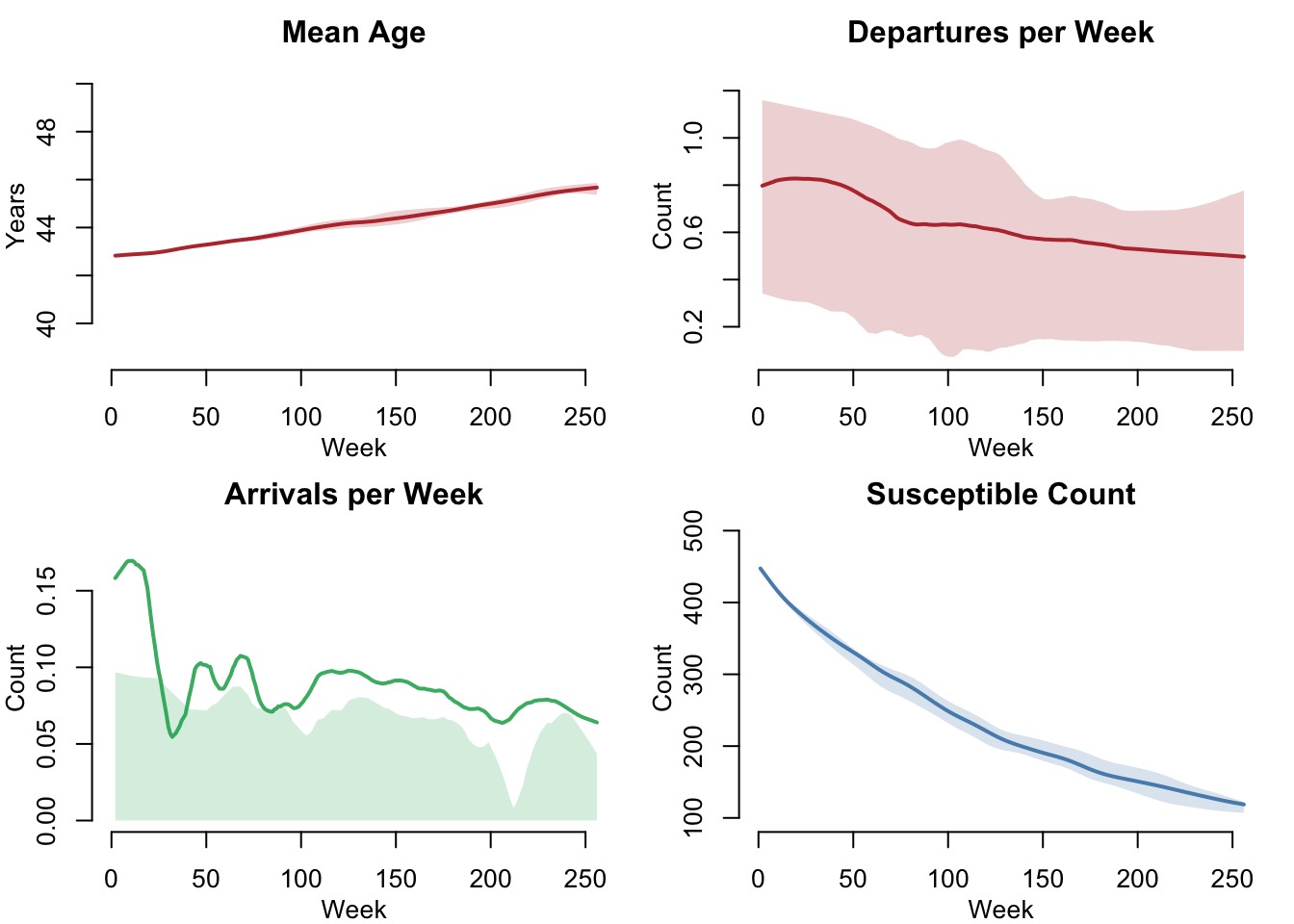
New Infections
par(mfrow = c(1, 1), mar = c(3, 3, 2, 1), mgp = c(2, 1, 0))
plot(sim_base, y = "si.flow",
main = "New Infections: Baseline vs. Disease Mortality",
ylab = "New Infections per Week", xlab = "Week",
mean.col = "steelblue", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "steelblue", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE)
plot(sim_lethal, y = "si.flow",
mean.col = "firebrick", mean.lwd = 2, mean.smooth = TRUE,
qnts.col = "firebrick", qnts.alpha = 0.2, qnts.smooth = TRUE,
legend = FALSE, add = TRUE)
legend("topright", legend = c("Baseline (mult=1)", "Lethal (mult=50)"),
col = c("steelblue", "firebrick"), lwd = 2, bty = "n")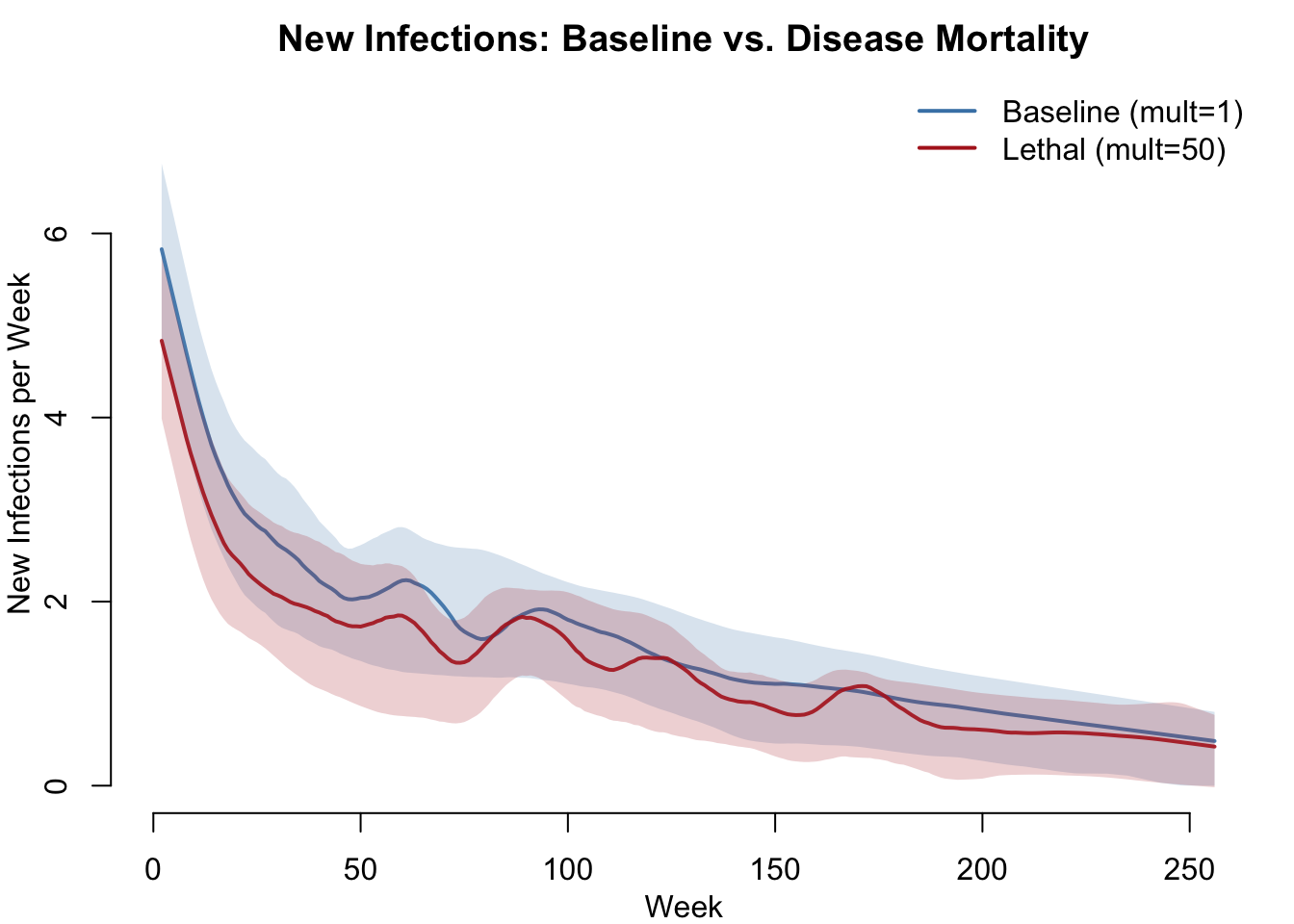
Summary Table
df_base <- as.data.frame(sim_base)
df_lethal <- as.data.frame(sim_lethal)
knitr::kable(data.frame(
Metric = c("Mean prevalence",
"Final prevalence",
"Mean population size",
"Final population size",
"Cumulative departures",
"Cumulative infections"),
Baseline = c(
round(mean(df_base$prev, na.rm = TRUE), 3),
round(mean(df_base$prev[df_base$time == max(df_base$time)], na.rm = TRUE), 3),
round(mean(df_base$num, na.rm = TRUE)),
round(mean(df_base$num[df_base$time == max(df_base$time)], na.rm = TRUE)),
round(mean(tapply(df_base$d.flow, df_base$sim, sum, na.rm = TRUE))),
round(mean(tapply(df_base$si.flow, df_base$sim, sum, na.rm = TRUE)))
),
Lethal = c(
round(mean(df_lethal$prev, na.rm = TRUE), 3),
round(mean(df_lethal$prev[df_lethal$time == max(df_lethal$time)], na.rm = TRUE), 3),
round(mean(df_lethal$num, na.rm = TRUE)),
round(mean(df_lethal$num[df_lethal$time == max(df_lethal$time)], na.rm = TRUE)),
round(mean(tapply(df_lethal$d.flow, df_lethal$sim, sum, na.rm = TRUE))),
round(mean(tapply(df_lethal$si.flow, df_lethal$sim, sum, na.rm = TRUE)))
)
))| Metric | Baseline | Lethal |
|---|---|---|
| Mean prevalence | 0.612 | 0.461 |
| Final prevalence | 0.865 | 0.674 |
| Mean population size | 500.000 | 424.000 |
| Final population size | 497.000 | 363.000 |
| Cumulative departures | 33.000 | 161.000 |
| Cumulative infections | 401.000 | 336.000 |
Parameters
Transmission
| Parameter | Description | Default |
|---|---|---|
inf.prob |
Per-act transmission probability | 0.15 |
act.rate |
Acts per partnership per week (built-in default) | 1 |
Vital Dynamics
| Parameter | Description | Default |
|---|---|---|
departure.rates |
Age-specific weekly mortality rates (86-element vector) | US life table data |
departure.disease.mult |
Multiplier on departure rates for infected individuals | 1 (baseline) or 50 (lethal) |
arrival.rate |
Per-capita weekly birth rate | mean(departure.rates) |
Network
| Parameter | Description | Default |
|---|---|---|
| Population size | Number of nodes | 500 |
| Mean degree | Average concurrent partnerships per node | 0.8 |
| Age assortativity | Mean absolute age difference within partnerships | 1.5 years |
| Partnership duration | Mean edge duration (weeks) | 60 (~1.2 years) |
Module Execution Order
aging → departures (dfunc) → arrivals (afunc) → resim_nets → infection → prevalenceAging runs first so that departure rates reflect updated ages. Arrivals follow departures to replace the departed. Network resimulation happens after demographic changes so the ERGM respects the new population composition. Infection then runs on the updated network.
Next Steps
- Add disease recovery to convert this SI model to an SIS or SIR model — see Adding an Exposed State for the progression module pattern
- Use more granular age categories or empirical life table data for country-specific mortality
- Add heterogeneous susceptibility that varies by age
- Add stage-dependent infectiousness and mortality — see the HIV model for disease stage–specific departure rates