Converts a transmission matrix from the get_transmat
function into a phylo class object.
Arguments
- x
An object of class
transmat, the output fromget_transmat().- vertex.exit.times
Optional numeric vector providing the time of departure of vertices, to be used to scale the lengths of branches reaching to the tips. Index position on vector corresponds to network id. NA indicates no departure, so branch will extend to the end of the tree.
- ...
Further arguments (unused).
Details
Converts a transmat() object containing information about the
history of a simulated infection into a ape::phylo object
representation suitable for plotting as a tree with
ape::plot.phylo(). Each infection event becomes a 'node'
(horizontal branch) in the resulting phylo tree, and each network
vertex becomes a 'tip' of the tree. The infection events are labeled with the
vertex ID of the infector to make it possible to trace the path of infection.
The infection timing information is included to position the phylo-nodes,
with the lines to the tips drawn to the max time value +1 (unless
vertex.exit.times are passed in it effectively assumes all vertices
are active until the end of the simulation).
If the transmat contains multiple infection seeds (there are multiple
trees with separate root nodes), this function will return a list of class
multiPhylo, each element of which is a phylo object. See
ape::read.tree().
Examples
# \donttest{
set.seed(13)
# Fit a random mixing TERGM with mean degree of 1 and mean edge
# duration of 20 time steps
nw <- network_initialize(n = 100)
formation <- ~edges
target.stats <- 50
coef.diss <- dissolution_coefs(dissolution = ~offset(edges), duration = 20)
est <- netest(nw, formation, target.stats, coef.diss, verbose = FALSE)
#> Starting simulated annealing (SAN)
#> Iteration 1 of at most 4
#> Finished simulated annealing
#> Starting maximum pseudolikelihood estimation (MPLE):
#> Obtaining the responsible dyads.
#> Evaluating the predictor and response matrix.
#> Maximizing the pseudolikelihood.
#> Finished MPLE.
# Parameterize the epidemic model as SI with one infected seed
param <- param.net(inf.prob = 0.5)
init <- init.net(i.num = 1)
control <- control.net(type = "SI", nsteps = 40, nsims = 1, verbose = FALSE)
# Simulate the model
mod1 <- netsim(est, param, init, control)
# Extract the transmission matrix
tm <- get_transmat(mod1)
head(tm, 15)
#> # A tibble: 15 × 8
#> # Groups: at, sus [15]
#> at sus inf network infDur transProb actRate finalProb
#> <int> <int> <int> <int> <dbl> <dbl> <dbl> <dbl>
#> 1 2 7 31 1 3 0.5 1 0.5
#> 2 2 94 31 1 3 0.5 1 0.5
#> 3 4 20 31 1 5 0.5 1 0.5
#> 4 4 33 94 1 2 0.5 1 0.5
#> 5 5 34 94 1 3 0.5 1 0.5
#> 6 5 72 33 1 1 0.5 1 0.5
#> 7 6 6 34 1 1 0.5 1 0.5
#> 8 6 36 72 1 1 0.5 1 0.5
#> 9 6 95 33 1 2 0.5 1 0.5
#> 10 7 40 34 1 2 0.5 1 0.5
#> 11 8 1 95 1 2 0.5 1 0.5
#> 12 8 48 7 1 6 0.5 1 0.5
#> 13 8 60 40 1 1 0.5 1 0.5
#> 14 8 68 95 1 2 0.5 1 0.5
#> 15 8 89 6 1 2 0.5 1 0.5
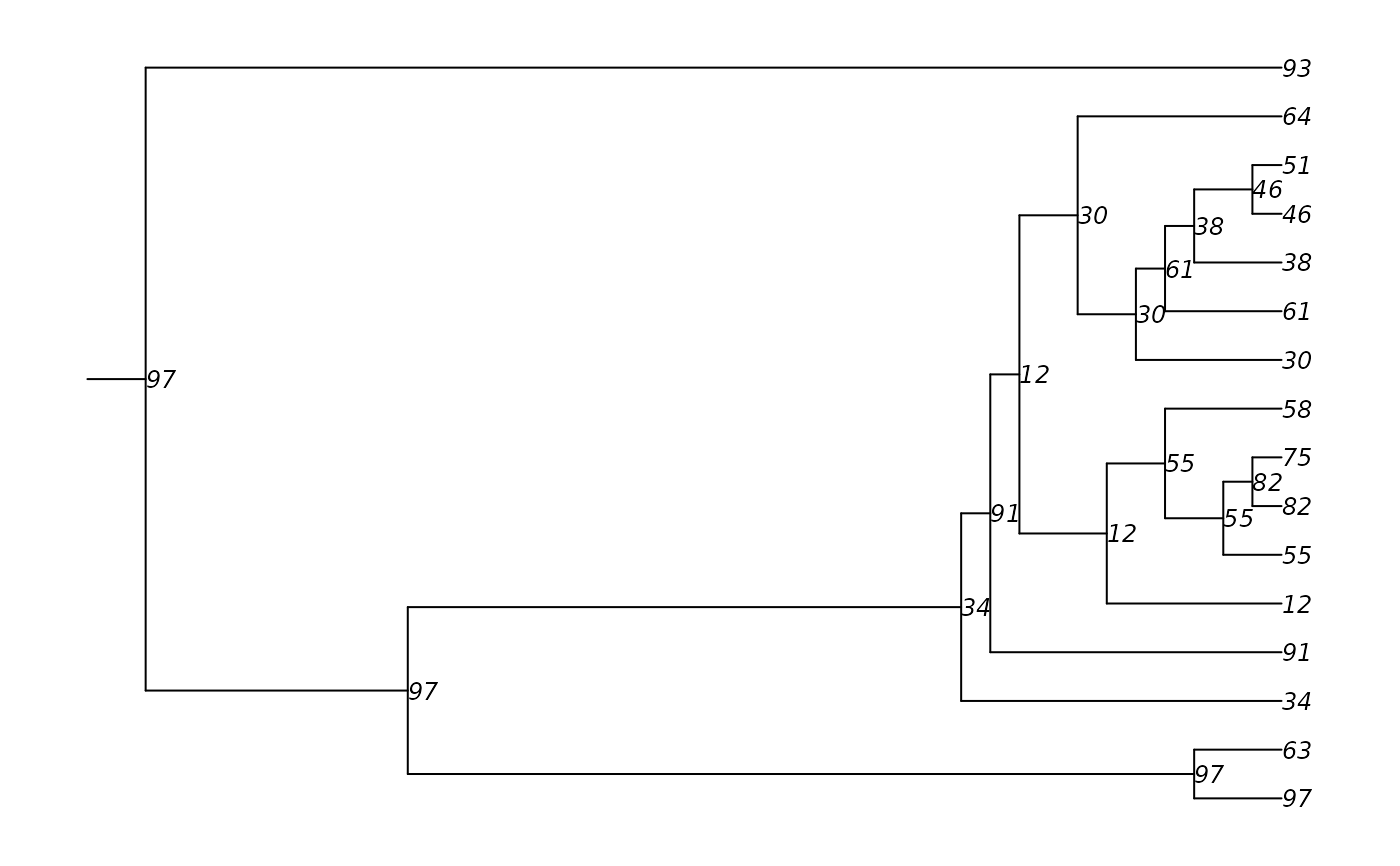
# Convert to phylo object and plot
tmPhylo <- as.phylo.transmat(tm)
par(mar = c(1,1,1,1))
plot(tmPhylo, show.node.label = TRUE,
root.edge = TRUE,
cex = 0.75)
 # }
# }